
Commentary On SwissMTB: Establishing Comprehensive Molecular Cancer Diagnostics In Swiss Clinics
Franziska Singer1,2, Daniel J. Stekhoven1,2*
1NEXUS Personalized Health Technologies, ETH Zurich, Otto-Stern-Weg 7, 8093, Zurich, Switzerland
2SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland
Introduction
High-throughput sequencing-based molecular diagnostics are an emerging practice in hospitals worldwide and hold great promises for personalized cancer treatment. In our recent publication, we highlight the importance of integrating such comprehensive sequencing and analysis strategies into clinical decision making1. We present the Swiss Molecular Tumor Board (SwissMTB), a workflow from biopsy to clinical reporting for molecular tumor boards and the results of the corresponding pilot study in Switzerland. In this commentary, we discuss the opportunities and challenges of comprehensive molecular cancer diagnostics, its promises for cancer treatment, and the lessons learned from SwissMTB.
SwissMTB is an attempt to bridge the gap between research and clinical routine for the personalized treatment of cancer patients. We implemented a reproducible workflow that supports treatment decisions by comprehensively analyzing the genetic landscape of tumors to link actionable genetic variants with therapy options (Figure 1). In a prospective pilot study together with the University Hospital Zurich, we collected tumor biopsies that underwent whole-exome sequencing (WES), whole-genome sequencing (WGS) and RNA sequencing (RNA-seq). Based on this high-throughput data, actionable genetic variants were identified and further prioritized based on their clinical significance. Focussing on treatment recommendations for the treating clinician, the respective findings were summarized in a clinical report and subsequently discussed in a molecular tumor board. In addition, we retrospectively analyzed data from tumor biopsies that were sequenced based on diverse gene panels at the University Hospital Basel.
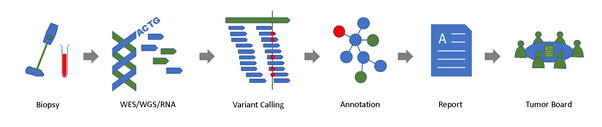
Figure 1: SwissMTB molecular diagnostics workflow.
DNA and RNA is extracted from a tumor biopsy (and paired control tissue, e.g., blood) and sequenced. The resulting data is analyzed to detect genetic alterations in the tumor sample, which are associated with potential therapy options. Suitable therapies and clinical trial opportunities are summarized in a clinical report, which is returned to the clinician and discussed in the molecular tumor board.
SwissMTB identified actionable targets and resistance-causing variants in 86% of the analyzed 22 patients. Eleven patients were analyzed before a treatment decision was taken. In five of these patients (45%), SwissMTB findings influenced treatment decisions. In the retrospective analysis, our workflow considerably enriched the information originally provided by the pathology report. Most noteworthy was the identification of resistance variants in two patients for treatments that indeed proved ineffective in the clinical follow-up. It is likely that a non-beneficial therapy could have been avoided, thus preventing an unnecessary treatment burden for the patients and saving the healthcare system needless costs.
Benefits of Comprehensiveness
Our pilot study shows that comprehensive sequencing for molecular diagnostics outperforms the popular but much smaller gene panels and the traditional Sanger sequencing of one or two specific targets. The complex changes and mechanisms driving cancer development2,3 cannot be fully captured by a limited set of hot spot genes. Admittedly, identifying a variant using a gene panel almost certainly will lead to a useful treatment decision, because these panels are composed according to prior knowledge on actionable mutations and their usefulness as predictive biomarkers. However, rare cancers and those with yet unknown drivers are rarely covered in the limited set of selected hot spot genes. Extending the scope beyond a priori defined panels will ensure that novel insights can be included continuously - now and in the future.
The seemingly prohibitive costs are a common argument against using comprehensive sequencing in clinical routine. However, this cost calculation needs to be put into the context of three aspects. First, avoiding treatments potentially lacking benefit will save costs outweighing those of a comprehensive sequencing protocol. Already in our small pilot study such cases do occur. Second, the use of comprehensive data extends beyond the specific treatment decision. Future insights can be validated in combination with the patient’s follow-up, further improving the cost-effectiveness of treatment. In addition, new biomarkers and actionable targets can be identified. Third, sequencing costs have decreased considerably in the last years4 and it can be expected that this trend carries on. Already today the focus of molecular profiling shifted from Sanger sequencing to gene panels, which are a well-integrated part in hospitals all over the world5-7.
We showcase this in SwissMTB and expect that the technologies will further shift to be even more comprehensive. First of all, important clinical features like the tumor mutational burden or the HLA-type can only be assessed reliably using WES or WGS. Although gene panels try to extrapolate the mutational burden, it will always remain a prediction. In addition, only comprehensive approaches allow to determine also mutational signatures8,9. These signatures provide valuable insights in the development of a tumor and may eliminate the need for further genetic testing, e.g. to determine the cancer type in case of an unknown primary tumor. Furthermore, on average we found a higher number of actionable variants per patient using comprehensive sequencing approaches. Finally, we could demonstrate that RNA-seq as a complementary technique not only verified genomic findings, but also guided recommendations by providing decisive information on fusion events and abnormal gene expression, especially in situations where either no point mutation could be identified or the mutations translate to previously used treatments. This reinforces the relevance of RNA-seq for clinical decision making, as in particular fusions and translocations considerably influence the genetic makeup of tumors10. Early phase clinical trials showed that cancers driven by gene fusions are actionable11. Thus, conventional methods like panel sequencing would fail to detect the potentially actionable targets of such fusion driven cancers.
Challenges of Data Interpretation
The wealth of information provided by high-throughput technologies comes at a price, as it has to be interpreted and prioritized for the subsequent use in the molecular tumor boards at the hospital, where typically only a limited amount of time per case is available. Reducing the list of possible targets to the most promising ones has a considerable influence on the time and effort necessary to formulate a concise clinical report and reach a treatment decision. Currently, this task is still one of the greatest challenges for diagnostic workflows, as it is not yet fully automated and requires manual inspection of sometimes large numbers of drug-target combinations. Given the aspired short turnaround time to provide information to a molecular tumor board, further work is needed to automate the retrieval of supportive or conflicting evidence from the literature. In SwissMTB we implemented a two-step procedure to achieve a trade-off between small turnaround time and finalized clinical report. Approximately one week after the biopsy we deliver a preliminary report containing all findings that passed the automated filtering and clinical annotation process. This enables our clinical partners to act on specific aspects, such as high mutational burden or a particular genetic variant, at an early stage. The final report including manual inspection is provided after approximately four weeks. In urgent cases, this three week-difference decides whether the diagnostic results can be used for a treatment decision. For the end-of-treatment line patients included in our study this two-step strategy proved beneficial several times.
The need to overcome the aforementioned challenge of reducing the list of possible targets to identify the truly relevant ones for clinical decision making in a limited amount of time, may lead to missing important targets or to falling back on only considering the typical set of a-priori candidates. In addition, the evaluation of interesting variants is always limited by the knowledge of the manual curator, or the knowledge accessible in public databases. This includes the availability of drug targets, which is currently the main reason that the list of identified variants is reduced to only few options that are actionable. It is to be expected that extended automation and improved algorithms for clinical interpretation, as well as increased global data sharing, will improve our ability to reduce the turnaround time while maintaining accuracy. Further, the growing number of known drug targets will increase the proportion of clinically relevant results from comprehensive sequencing approaches.
Another important part of clinical interpretation is linking potential treatments to the health regulations in a particular country. SwissMTB is unique in its attempt to integrate information from the Swiss authorities regarding approval of treatments. This information is critical in order to decide whether a potential treatment is feasible for a patient, particularly in regard to costs covered by health insurances. We argue that molecular tumor board initiatives always need to account for the regulations in their respective country, in order to provide meaningful interpretation and summary for the treating clinician. We even go one step further in SwissMTB and directly link potential therapies to recruiting clinical trials in and around Switzerland. Even though a therapy might not be approved yet, it might be accessible via such trials.
Summary
SwissMTB combines a robust and reproducible workflow for the analysis of comprehensive molecular data with sophisticated functionality to annotate and clinically interpret actionable targets. Once the benefits of using comprehensive technologies have been established, these approaches will likely become standard diagnostics, not only for patients that have progressed on standard therapies but for every cancer patient entering the clinic. Our molecular diagnostics workflow provides a prototype that can form the basis for streamlined profiling and reporting required to enable such routine clinical use in Switzerland.
References
- Singer F, Irmisch A, Toussaint NC, et al. SwissMTB: establishing comprehensive molecular cancer diagnostics in Swiss clinics., BMC Med Inform Decis Mak. 2018; 18: 89. doi:10.1186/s12911-018-0680-0.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646–674. doi:10.1016/j.cell.2011.02.013.
- Sabarinathan R, Pich O, Martincorena I, et al. The whole-genome panorama of cancer drivers. BioRxiv. 2017. doi:10.1101/190330.
- Wetterstrand KA, DNA Sequencing Costs, DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). (n.d.). http://www.genome.gov/sequencingcostsdata (accessed December 19, 2018).
- Shin SH, Bode AM, Dong Z. Addressing the challenges of applying precision oncology. Npj Precision Onc. 2017; 1: 28. doi:10.1038/s41698-017-0032-z.
- Hamblin A, Wordsworth S, Fermont JM, et al., Clinical applicability and cost of a 46-gene panel for genomic analysis of solid tumours: Retrospective validation and prospective audit in the UK National Health Service. PLoS Med. 2017; 14: e1002230. doi:10.1371/journal.pmed.1002230.
- Tsongalis GJ, Peterson JD, de Abreu FB, et al. Routine use of the Ion Torrent AmpliSeqTM Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med. 2014; 52: 707–714. doi:10.1515/cclm-2013-0883.
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500: 415–421. doi:10.1038/nature12477.
- Soh KP, Szczurek E, Sakoparnig T, et al. Predicting cancer type from tumour DNA signatures. Genome Med. 2017; 9: 104. doi:10.1186/s13073-017-0493-2.
- Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018; 23: 227–238.e3. doi:10.1016/j.celrep.2018.03.050.
- Groisberg R, Hess KR, Hong DS, et al. Outcomes of patients with gene fusion driven cancers treated on early phase clinical trials. J Clin Oncol. 2018; 36: 2590–2590. doi:10.1200/JCO.2018.36.15_suppl.2590.