
Epigenetic aberration at enhancer regions in gastric cancer
Atsushi Okabe1, Atsushi Kaneda1*
1Department of Molecular Oncology, Graduate School of Medicine, Chiba University, Chiba, Japan
Abstract
Cancer arises through the accumulation of genetic and epigenetic alterations. Comprehensive analyses of human cancer epigenomes over the past decade have revealed that chromatin and epigenetic aberrations induced by genetic, metabolic, and environmental stimuli play important roles in tumor initiation as well as progression. Among these aberrations, DNA hypermethylation at promoter regions is one of the major mechanisms to silence tumor suppressor genes in cancer, and has been studied in detail. For gastric cancer, for example, we and other groups have conducted genome-wide DNA methylation analyses, and classified gastric cancer into several DNA methylation epigenotypes. Gastric cancer with Epstein-Barr virus (EBV) infection exhibits the most extensive hypermethylation phenotype among all the human malignancies, and EBV infection itself is shown to cause aberrant DNA methylation induction. EBV infection also alters histone modifications, not only at promoter regions but also at enhancer regions. Epigenetic alteration at enhancers causes aberrant regulation of cancer-related genes together with epigenetic alteration at promoters, and it is known to contribute to tumorigenesis. We here review epigenetic aberration at enhancer regions in gastric cancer.
Introduction
For gastric cancer, we and other groups have conducted integrative genomic and genome-wide DNA methylation analyses, and classified gastric cancer into several molecular subtypes1–4. Gastric cancer with Epstein-Barr virus (EBV) infection were characterized by frequent ARID1A mutation, a lack of TP53 mutation and relatively chromosome stable. In addition, it shows the highest degree of hypermethylation among all the human malignancies but had minimal demethylation3,4, and EBV infection itself is shown to cause aberrant DNA methylation induction2. We recently showed that EBV infection also alters histone modifications, not only at promoter regions but also at enhancer regions5,6 and enhancer alteration is known to contribute to tumorigenesis through aberrant expression of cancer related genes7.
Transcriptional regulation by enhancers
Enhancers are cis-acting DNA regulatory elements that increase the transcriptional output of target genes to regulate cell-type or tissue-type specific genes during development and differentiation. Enhancer sequences contain short DNA motifs that transcription factors can bind to in a sequence-specific manner. These binding proteins exclude nucleosome, recruit epigenetic modifiers, and stabilize chromatin loops between their target regions by architectural proteins.
Nucleosomes in the vicinity of active enhancers typically contain specifically modified histones. Histone H3 lysine 4 mono-methylation (H3K4me1) is predominant at the enhancers8 but H3K4 tri-methylation (H3K4me3) is predominant at the promoters. H3K4 di-methylation (H3K4me2) is enriched around H3K4me3 modification but also observed at enhancers. The activity of promoters and enhancers is determined by modification at H3 lysine 27 (H3K27). Whereas H3K27 is tri-methylated at inactive/poised enhancers and promoters, H3K27 is acetylated at active enhancers and promoters9–11. In mammals, MLL3 and MLL4 constitute major H3K4 monomethyl-transferases and also function with Utx, a subunit of the complex, as an enhancer-specific H3K27 demethylase against tri-methylated H3K27 (H3K27me3)12. Acetylated H3K27 (H3K27ac) are regulated by histone acetyl transferases such as p300 and CBP. Enhancer-bound transcription factors interact with the basal transcription machinery on promoters, through the Mediator complex and transcription elongation complex including BRD4 (Figure 1). The architectural proteins, e.g., CTCF and cohesin, and their loading factors such as NIPBL are involved in stabilizing these long-range interactions13–16.
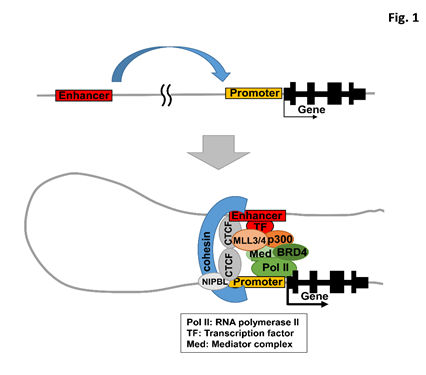
Figure 1: A model for transcriptional activation by enhancer.
TF, transcription factor. Med, Mediator complex. Pol II, RNA polymerase II. Transcription factors often recruit chromatin-modifying coactivators, transcription elongation complex, and architectural proteins in interaction between enhancer and promoter.
Enhancer dysregulation in cancer
Recent studies have shown that single-nucleotide polymorphisms, insertions, or deletions induce aberrant activation or repression at enhancers and alter the gene expression17,18. Also, the MLL3 and MLL4 genes have been reported to be frequently mutated in many different forms of cancer, some of which include bladder cancer, breast cancer, liver cancer, gastric cancer, etc19–22. These suggested that enhancer regions are important to maintain the tissue-specific expression and that the enhancer dysregulation leads to diseases including cancer.
Super-enhancers are large clusters of enhancers that define cell identity by upregulating neighboring genes. Super-enhancers associated with oncogenic driver genes are aberrantly activated in cancer cells through many different mechanisms23. The genetic mechanisms of super-enhancer acquisition in cancer include DNA translocation24–26, focal amplification23,27,28, and nucleation by small insertions/deletions that create master transcription factor binding sites29. Additional epigenomic mechanisms of super-enhancer formation in cancer are oncogenic overexpression of transcription factors23, oncogenic fusion of transcription factors such as EWS-FLI30, and the consequences of upstream oncogenic signaling such as RAS-dependent signaling to chromatin31 (Figure 2).
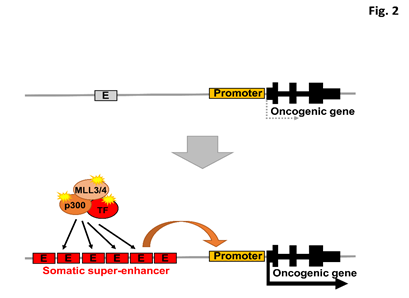
Figure 2: Model of somatic super-enhancer activation in cancer.
E, enhancer. TF, transcription factor. Dysregulated transcription factors and epigenetic modifiers, and somatic super-enhancers acquired through genomic structural variations, e.g. focal amplification of an enhancer element, activate driver oncogenes.
In primary gastric adenocarcinoma, Ooi et al. identified somatic super-enhancers using clinical tissue samples, which mainly include genomically stable and chromosomally instable gastric cancer subtypes with lower DNA methylation, and not microsatellite instable and EBV(+) subtypes with higher DNA methylation32. They experimentally confirmed direct binding of transcription factors CDX2 and HNF4A at super-enhancers aberrantly identified in gastric cancer, and silencing of these transcription factors could repress the somatic super-enhancers.
Enhancer dysfunction is also related to metastasis and patient mortality. Bell et al. performed an integrative analysis of DNA methylation, RNA-seq, and small RNA-seq profiles in thousands of patients, including 25 diverse primary malignancies and seven body sites of metastatic melanoma33. They identified differentially methylated regions in enhancers (eDMR); eDMR enabled classification of primary tumors according to physiological organ systems, and correlated with patient outcome. Roe et al. reported that metastasis is promoted through enhancer reprogramming in pancreatic cancer34. They used an organoid culture system to investigate how transcription and the enhancer landscape are altered during discrete stages of disease progression in a mouse model of pancreatic ductal adenocarcinoma. They showed that the cancer cells become more invasive and less anchorage-dependent for growth through FOXA1-dependent enhancer reprogramming in vitro, as well as more metastatic in vivo.
Enhancer dysregulation in EBV(+) gastric cancer
Recently, we provided evidence that EBV infection in gastric epithelial cells induces alteration of global histone modification, including active and repressive histone marks6. We showed that de novo DNA methylation at promoter regions is correlated with low level or decrease of active histone marks (H3K4me3 and H3K27ac)5. H3K27me3 loss at promoter regions were replaced by de novo DNA methylation and expression of associated genes were maintained repressed5,6. In addition, de novo DNA methylation was concomitantly observed at regions with increase of H3 lysine 9 tri-methylation (H3K9me3)6, in agreement with previous findings about association of H3K9me3 with DNA methylation35.
On the other hands, loss of repressive marks (H3K27me3 or H3K9me3) and concomitant enhancer activation through an increase of H3K4me1 and H3K27ac dynamically occurred. Such enhancer activation might contribute to tumorigenesis by activating neighboring oncogenic genes6. We also showed that enhancer repression through deacetylation of H3K27ac and induction of DNA methylation might contribute to tumorigenesis by repressing neighboring tumor suppressor genes. We identified activated enhancers during EBV infection, with Runx1 and Ets1 motifs enriched at these regions. In addition, we showed that the GATA3 and GRHL1 motifs were enriched at repressed enhancers6.
Host and viral factors that associated with epigenetic alteration may facilitate the initiation, maintenance, and evolution of EBV(+) gastric cancer. Ten-eleven-translocation (TET) proteins might function as resistant factors against de novo DNA methylation, to protect unmethylated status at promoter region36. Downregulation of TET2 by EBV transcripts such as BARF0 or LMP2A36, and upregulation of Dnmt1 in EBV(+) gastric cancer2 are potential causes of epigenetic repression by DNA methylation induction. Moreover, EP300 was upregulated, and several histone deacetylases (HDACs) were downregulated during EBV infection in gastric epithelial cells6,36. These expression alterations were suggested to be involved in enhancer activation, and further investigations are necessary to fully clarify the molecular mechanism to induce host chromatin alteration in gastric epithelial cells.
Clinical application targeted enhancer regions
Preventing somatic super-enhancers or reactivating repressed tumor suppressive enhancers might be possible therapeutic strategies. Super-enhancers are especially vulnerable to inhibition of enhancer factors because of the cooperative features of enhancer components37. Genes encoding tumor cell master transcription factors acquire especially large super-enhancers, with exceptionally high densities of enhancer factors23. Therefore, super-enhancer-driven oncogenic transcription factors are vulnerable to inhibition of BRD438.
Six epigenetic agents are approved by the US Food and Drug Administration (FDA), including two DNA methyltransferase (DNMT) inhibitors (azacytidine and decitabine) and four HDAC inhibitors (vorinostat, romidepsin, belinostat, and panobinostat), as anticancer drugs. These may activate repressed regions including promoters and enhancers of tumor suppressor genes, but reprogram the epigenome broadly and randomly and thus affect non-oncogenic genes as well39,40. Therefore, a region-specific approach would be expected to reduce possible side effects.
Recently, CRISPR/Cas9 based technologies have been developed and enable us to perform site-specific editing of epigenome. Hilton et al. reported targeted acetylation using the nuclease-null dCas9 fusion to the p300 and successfully activated expression of genes associated with target promoters and enhancers in human HEK293T cells41. Morita et al. used dCas9-SunTag system to achieve efficient recruitment of an anti-GCN4 scFV fused TET1 catalytic domain42. They demonstrated targeted demethylation of CpGs in culture cells and in vivo in mouse fetuses. This dCas9-SunTag system was also applied to recruit DNMT3A for region-specific DNA methylation induction and repressed its target genes in HEK293T cells43.
These region-specific epigenetic editing technologies might be expected as a novel epigenetic therapeutic strategy. CRISPR/Cas9 based technologies, however, require transfection step, e.g., electroporation or viral infection system, in delivery into the nucleus. We and others have tried to overcome this point using a cell-permeable small molecule called pyrrole-imidazole polyamide, which binds to the minor groove of targeted DNA sequence. We reported that a pyrrole-imidazole polyamide could inhibit DNA methylation in a gene promoter region specifically around its targeting sequence, and inhibit silencing of the gene44. Pyrrole-imidazole polyamides can also be conjugated with inhibitors against histone modification enzymes to alter histone status at selective genomic regions45. These techniques to modify epigenomic status at selective regions might be helpful for the development of a new epigenetic therapeutic strategy.
Acknowledgement
This study was supported by the Japan Agency for Medical Research and Development (AMED; Practical Research for Innovative Cancer Control 17ck0106263h0001 to AK) and the Japan Society for the Promotion of Science (JSPS; 16H05412 to AK).
Conflicts of interest
All authors have no conflicts of interest to disclose.
References
- Kaneda A, Kaminishi M, Yanagihara K, et al. Identification of Silencing of Nine Genes in Human Gastric Cancers. Cancer Res. 2002; 62(22) :6645-6650.
- Matsusaka K, Kaneda A, Nagae G, et al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011; 71(23): 7187-7197.
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513(7517): 202-209.
- Wang K, Yuen ST, Xu J, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014; 46(6): 573-582.
- Funata S, Matsusaka K, Yamanaka R, et al. Histone modification alteration coordinated with acquisition of promoter DNA methylation during Epstein-Barr virus infection. Oncotarget. 2017; 8(33): 55265-55279.
- Okabe A, Funata S, Matsusaka K, et al. Regulation of tumour related genes by dynamic epigenetic alteration at enhancer regions in gastric epithelial cells infected by Epstein-Barr virus. Sci Rep. 2017; 7(1): 7924.
- Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014; 53(6): 859-866.
- Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007; 39(3): 311-8.
- Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459(7243): 108-112.
- Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010; 107(50): 21931-21936.
- Rada-Iglesias A, Bajpai R, Swigut T, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011; 470(7333): 279-83.
- Herz HM, Mohan M, Garruss AS, et al. Enhancer-associated H3K4 monomethylation by trithorax-related, the drosophila homolog of mammalian MLL3/MLL4. Genes Dev. 2012; 26(23): 2604-2620.
- Ren X, Siegel R, Kim U, et al. Direct Interactions of OCA-B and TFII-I Regulate Immunoglobulin Heavy-Chain Gene Transcription by Facilitating Enhancer-Promoter Communication. Mol Cell. 2011; 42(3): 342-355.
- Liu Z, Scannell DR, Eisen MB, et al. Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell. 2011; 146(5): 720-731.
- Koch F, Fenouil R, Gut M, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011; 18(8): 956-963.
- Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010; 467(7314): 430-435.
- Zhang X, Cowper-Sal-lari R, Bailey SD, et al. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res. 2012; 22(8): 1437-1446.
- Yao L, Tak YG, Berman BP, et al. Functional annotation of colon cancer risk SNPs. Nat Commun. 2014; 5: 5114.
- Ellis MJ, Ding L, Shen D, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012; 486(7403): 353-60.
- Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012; 44(7): 760-764.
- Gui Y, Guo G, Huang Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011; 43(9): 875-878.
- Zang ZJ, Cutcutache I, Poon SL, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012; 44(5): 570-574.
- Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155(4): 934-47.
- Drier Y, Cotton MJ, Williamson KE, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016; 48(3): 265-272.
- Gröschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in Leukemia. Cell. 2014; 157(2): 369-381.
- Northcott PA, Lee C, Zichner T, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014; 511(7510): 428-434.
- Shi J, Whyte WA, Zepeda-Mendoza CJ, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013; 27(24): 2648-2662.
- Zhang X, Choi PS, Francis JM, et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016; 48(2): 176-182.
- Mansour MR, Abraham BJ, Anders L, et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014; 346(6215): 1373-1377.
- Tomazou EM, Sheffield NC, Schmidl C, et al. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Rep. 2015; 10(7): 1082-1095.
- Nabet B, Ó Broin P, Reyes JM, et al. Deregulation of the Ras-Erk Signaling Axis Modulates the Enhancer Landscape. Cell Rep. 2015; 12(8): 1300-1313.
- Ooi WF, Xing M, Xu C, et al. Epigenomic profiling of primary gastric adenocarcinoma reveals super-enhancer heterogeneity. Nat Commun. 2016; 7: 12983.
- Bell RE, Golan T, Sheinboim D, et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res. 2016; 26(5): 601-611.
- Roe JS, Hwang C Il, Somerville TDD, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell. 2017; 170(5): 875-888.e20.
- Du J, Johnson LM, Jacobsen SE, et al. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015; 16(9): 519-532.
- Namba-Fukuyo H, Funata S, Matsusaka K, et al. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget. 2016; 7(49): 81512-81526.
- Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell. 2017; 168(4): 629-643.
- Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153(2): 320-334.
- Jones PA, Issa J-PJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016; 17(10): 630-641.
- Dawson MA. The cancer epigenome - concepts, challenges and theraputic opportunities. Science. 2017; 1(5): 379-385.
- Hilton IB, D’Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015; 33(5): 510-517.
- Morita S, Noguchi H, Horii T, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016; 34(10): 1060-1065.
- Huang YH, Su J, Lei Y, et al. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017; 18(1): 1-11.
- Shinohara K, Yoda N, Takane K, et al. Inhibition of DNA Methylation at the MLH1 Promoter Region Using Pyrrole–Imidazole Polyamide. ACS Omega. 2016; 1(6): 1164-1172.
- Han L, Pandian GN, Junetha S, et al. A synthetic small molecule for targeted transcriptional activation of germ cell genes in a human somatic cell. Angew Chemie - Int Ed. 2013; 52(50): 13410-13413.