
Pyrrole indolin-2-One Based Kinase Inhibitor as Anti-Cancer Agents
Ting-Hsuan Yang1,2, Ren-Jun Hsu2, Wen-Hsin Huang1, An-Rong Lee1*
School of Pharmacy, National Defense Medical Center, No. 161, Section 6, Mingchuan East Road, Taipei 11490, Taiwan
Tri-Service General Hospital Biobank, No.325, Sec. 2, Chenggong Rd. Taipei 11490, Taiwan
Abstract
Cancer cells are characterized by uncontrolled proliferation after escaping from inherent physiological constraints on growth and survival and by destructive invasion of the healthy surrounding tissues. Many kinases involved in signal transduction are overactive in malignant tumor cells. Thus, pharmacotherapeutic interventions targeting kinases responsible for signal transduction of cancer hallmarks have become promising in developing novel anticancer agents. Pyrrole indolin-2-one (or pyrrole oxindole, 1), a lead scaffold of kinase inhibitors, is used for anti-angiogenesis via inhibiting vascular endothelial growth factor receptors (VEGFs) and platelet-derived growth factor receptors (PDGFRs). The kinase selectivity and inhibitory activity of pyrrole indolin-2-one 1 can be significantly influenced through structural modifications. This mini-review provides a detailed overview of structural modification of pyrrole indolin-2-one derivatives for the development of novel kinase inhibitors.
Introduction
The activity of proteins or enzymes can be enhanced after phosphorylation by specific protein kinase. Protein phosphorylation is a fundamental mechanism of signal transduction used by all cells to regulate their protein properties in response to external or internal signals1. Phosphorylation and dephosphorylation regulates most cellular functions in both normal and abnormal cells2. For instance, kinase signal transduction is involved in many of cancer hallmarks such as stimulating cell proliferation, survival, and tumor induced angiogenesis, which is critical for supplying oxygen, nutrients, and paths of metastasis of tumor tissues3,4. Additionally, tumor-induced angiogenesis can be stimulated by many pro-angiogenesis growth factors, such as angiopoietin-2, epidermal growth factors (EGFs), fibroblast growth factors (FGFs), vascular endothelial growth factors (VEGFs), and platelet-derived growth factors (PDGFs)5. Among them, VEGFs, PDGFs, and their receptor tyrosine kinases (RTKs) play key roles in tumor angiogenesis signal transduction6. Therefore, most small molecule kinase inhibitors (SMKIs) can inhibit cancer cell proliferation by inhibiting RTKs of VEGFs and PDGFs, can block the downstream signal pathways, such as proliferation, migration, and enhance cell survival of endothelial cells, fibroblast, and vascular smooth muscle cells (Figure 1)7-10. Recently, several small-molecule anti-angiogenesis agents targeting VEGFRs and PDGFRs have been developed and approved for clinical use.
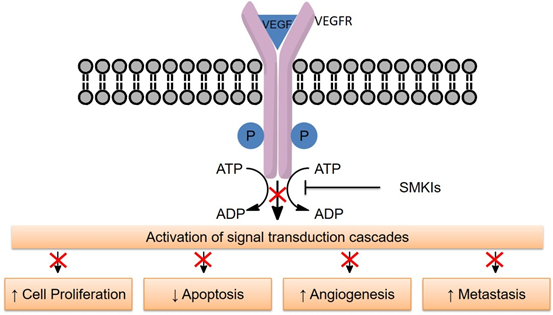
Figure 1: Small molecule kinase inhibitors (SMKIs) inhibit the proliferation of cancer cells by blocking the downstream signal pathways.
Pyrrole indolin-2-one 1 is known to be a critical structures in some inhibitors of receptor tyrosine kinases (RTKs). Semaxanib (SU5416, 2), a derivative of pyrrole indolin-2-one 1, was the first example of these tested clinically as a potent RTK inhibitor of VEGFRs and PDGERs for anti-angiogenesis, although not licensed for use11. Among the most potent anti-angiogenesis and anti-cancer agents is sunitinib (3), which is produced by structural modification of semaxanib and approved by the FDA for the treatment of renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumor (GIST) in 200611. Sunitinib was the first cancer drug simultaneously approved for two different indications. In 2011, sunitinib was approved by the FDA for treating patients with progressive neuroendocrine cancerous tumors located in the pancreas that cannot be removed by surgery or that have metastasized11. Sunitinib inhibits cellular signaling by specifically targeting multiple kinases, including VEGFRs, PDGFRs, aurora kinases (A, B and C), RET, Flt-3, Yes, Src, CSF-1R, and c-Kit12-17.
The ATP-binding site of the RTK can be divided into six regions: adenine pocket, specific pocket, hydrophobic pocket I, hydrophobic pocket II, ribose binding region, and phorphate binding region18. Since the ATP-binding sites of RTKs are highly conserved, most SMKIs targeting the ATP-binding site show a broad kinase inhibitory activity. Therefore, structural modification of small molecule RKTs has become a very important strategy for improving the selectivity and activity of kinase inhibition19. Structure-activity relationships (SARs) of the pyrrole indolin-2-one derivatives have been comprehensively investigated in previous works20-31. Both the crystal data and molecular modeling data demonstrate that the C(3’) amino tail of pyrrole indolin-2-one derivatives is exposed to the solvent region of the ATP-binding sites24,28-30,32,33 and has a critical influence on the solubility of the pyrrole indolin-2-one derivatives33,34. Additionally, the C(3’) amino tail can be further modified to target the pyrrole indolin-2-ones to some specific cell, organelle, or protein, such as mitochondria35-38. The oxindole moiety (herein oxindole means the C(2) of indole is oxidized), which is the key fragment in the binding affinity of kinases and pyrrole indolin-2-ones, occupies the adenine pocket, forming two hydrogen bonds with kinases4,22,30,39. Finally, appropriate substitutions of the oxindole moiety can enhance affinity by increasing kinase interactions4.
This mini-review provides an overview of the development of SMKIs based on structural modification of the pyrrole indolin-2-one scaffold 1 (Figure 2).
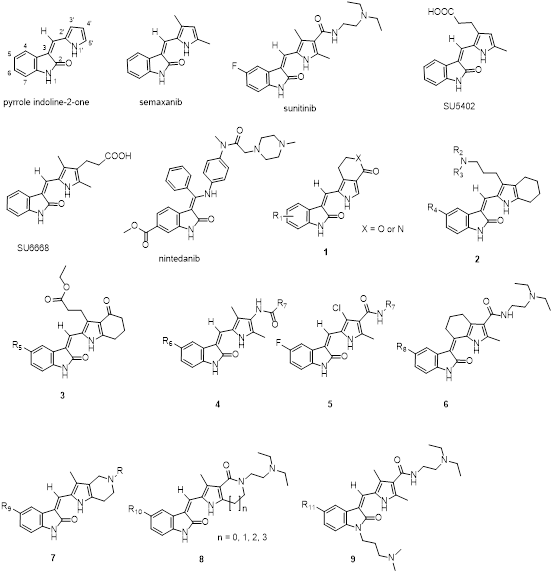
Figure 2: Structures of small molecule kinase inhibitors (SMKIs) based on structural modification of the pyrrole indoline-2-one scaffold.
Modification of Pyrrole Moiety
The pyrrole moiety of the pyrrole indolin-2-one scaffold 1 binds to the hydrophobic pocket I of the ATP-binding site of kinases, which is also known as the selective pocket18,40. Modification of the pyrrole moiety of the pyrrole indolin-2-ones, therefore, can positively affect kinase inhibition selectivity. First, lactamization or lactonization of C(3’) and C(4’) of the pyrrole of pyrrole indolin-2-ones produced compounds 5 and were found to change the selectivity of cyclin-dependent kinases (CDK)31. Compared to semaxanib (SU5416, 2) and SU5402 (4), the tetrahydroindole-containing 6 have stronger Src inhibiting effects and a broad range of VEGR-2 and PDGFRβ inhibiting effects. Obviously, the scaffold of compounds 6 is a possible structure unit in the determination of the activity and selectivity of Src inhibition17.
Fusing the pyrrole moiety with 4’-oxo-4’,5’,6’,7’-tetrahydroindole and introducing a carboxyethyl group to the side chain attached on C(3’) obtains compounds 7, which increase inhibitory activities of Aurora A and Aurora B41. Changing the carboxamide bond of sunitinib (3) through bioisosterism (amide-acyl bond) resulted in compounds 8 which exhibit a great enhancement in FLT3 and VEGFR-2 selectivity42,43. Changing the C(3’)-methyl of sunitinib (3) into chloride (e.g., 9) increases antitumor activities and reduces cardiotoxicity33. Compared to sunitinib (3), further modification by fusing a six-membered ring to the pyrrole ring led to the discovery of compounds 10, which show similar inhibitory activity against PDGFRβ but more potent to VEGFR-2 and c-Kit30. Fusing the pyrrole moiety of semaxanib (2) with a 1,4,6,7-tetrahydro-pyrrolo[3,2-c]pyridine results in compounds 11, which have various inhibitory activities in VEGFR-2, ALK, PDGFRα, and RET, and specifically selective to LRRK2 inhibition23.
Additionally, fusing the pyrrole moiety with five-, six-, seven-, eight-membered heterocycle (e.g., 12) specifically increases the kinase inhibitory activity and selectivity28,29,44. Interestingly, the smaller hetercyles (five- or six-membered heterocycle) fused pyrrole indolin-2-ones show much better VEGFR and PDGFRβ inhibitory activity27,28,43. Compared to sunitinib (3), the five- and six-membered heterocycle fused pyrrole indolin-2-ones demonstrate better inhibiting activity against both VEGFR-2 and PDGFRβ28,29,44. Among them, famitinib (n = 1, R11: -F in 12) showed significantly improved progression free survival (PFS) in patients with advanced colorectal cancer in phase IIb study, while its toxicity was manageable45.
Since the ATP-binding site is highly conserved, most small molecule kinase inhibitors have various kinase inhibiting activities. Therefore, two or more kinase targets can be simutaneously inhibited by the same molecule, i.e., multitarget kinase inhibitor. However, varied kinase inhibitory activity also leads to toxicities or side effects, such as hypertension and cardiotoxicity45. Thus, improving the inhibitory selectivity and/or reducing toxicities of theraputic tagets should be priortized when developing SMKIs. Data recently reported in the literature indicate that kinase inhibitory selectivity is closely related to the structural modification in the pyrrole moiety in this scaffold. Recent research shows that replacing the pyrrole moiety into other aromatic rings also influences kinase inhibitory selectivity. For instance, nintedanib (13) received FDA approval for use in targeting VEGFR, FGFR, and PDGFR in the treatment of non-small-cell lung cancer (NSCLC) in 201446.
Structural Modification of Oxindole Moiety
The oxindole moiety of pyrrole indolin-2-ones can provide two hydrogen bonds, which are critical for the binding of pyrrole indolin-2-ones to the ATP-binding site of the kinases, i.e., VEGFR-2, FGFR-1, and PDK-14,22,23,28-31,39,42,47. The C(5) and C(6) positions of the pyrrole indolin-2-ons are considered as one of most effective positions for interaction with the ATP-binding site20-30,48. Since substitutions on C(5) and C(6) could substantially affect ligand protein binding affinity, these sites of the oxindole moiety are the most commonly selected for structural modification.
Modification on N(1)
Prodrugs of semaxanib (2) and sunitinib (3) are usually generated by linking squalene to their N(1) positions49. The squalenoyl prodrugs can form nanoparticles and can be activated by changing pH in the body49. Another example of the sunitinib prodrug is N-hydroxylmethylsunitinib (AST-004), which is activated via hydrolysis catalyzed with esterase50. This prodrug can also reduce the toxicity of sunitinib (3)50. Introducing a 3-dimethylaminopropyl to N(1) of the pyrrole indolin-2-ones led to the discovery of compounds 14 which display various anti-tumor activities51.
Modification on C(5) and C(6)
Replacement on C(5)-halogen is the most common modification of the oxindole moiety of the pyrrole indolin-2-one derivatives, which have excellent overall inhibiting activities against VEGFR-2 and PDGFRβ22,28,33,34,42-44,52. The C(5)-halides form hydrophobic interaction between the ligand and the hydrophobic pocket II of the ATP-binding site. However, bioisosteric replacement of C(5)-halogens with C(5)-CF3 does not improve inhibition activity against VEGFR-2 and PDGFRβ39,41. Linking the alkyl groups or aromatic rings with sulfonamides, amides, or a urea bond to C(5) or C(6) is very common in the modification on the oxindole moiety18,21,23,27-29,42. The products provide the best possible activity by enabling additional hydrogen bonding interactions. Alkyl or aromatic groups might also enable further hydrophobic interactions with the hydrophobic pocket II of the ATP binding site. The effect of a methoxy group at C(5) of the oxindole moity on kinase inhibitory activity and selectivity is highly dependent on the C(3) substituents of indolin-2-one23,47,52. Introducing a -OH or a -SH to the C(5) position of the oxindole moity of the five-membered heterocycle fused pyrrole indolin-2-ones significantly improves inhibitory activity against VEGFR-2 and PDGFRβ28.
The pyrrole indolin-2-one derivatives can be turned into irreversible kinase inhibitors by introducing a chloromethylketone, a chloroacetamide, or other Michael acceptors to generate Nek2 selective inhibitors24.
In summary, pyrrole indolin-2-one analogues are most promising in kinase inhibitor development. Among the structural modifications, pyrrole indolin-2-ones with substitution of halides on C(5), such as sunitinib (3), approved by the FDA, and famitinib (n = 1, R11: -F in 12), tested in phase III clinical trial for colorectal cancer treatment, possess superior inhibitory activity against VEGFR-2 and PDGFRβ53. Apart from C(5)-halide substituents, the kinase selectivity and activity of other C(5) or C(6) substituted (e.g., -OMe, -OH, COOMe, -SH) pyrrole indolin-2-ones depend on the modification of the pyrrole moiety. It is noted, however, that some adverse reactions, such as hypertension and cardiotoxicity, were still observed in clinical trials45. To improve the safety and promote the quality of life of patients, further investigation for novel compounds with better activity/selectivity and lower toxicity is needed.
Acknowledgement
The authors would like to thank the Ministry of Science and Technology, R.O.C. for financially supporting this research under Contract No. MOST104-2320-B-016-004.
References
- Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep. 2011; 1: 90.
- Cohen P. Protein Kinases — the major drug targets of the twenty-first century. Nat Rev Drug Disc. 2002; 1: 309-315.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5): 646-674.
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009; 9(1): 28-39.
- Tonini T, Rossi F, Claudio PP. Molecular basis of angiogenesis and cancer. Oncogene. 2003; 22(42): 6549-6556.
- Roskoski R. Jr. Sunitinib: a VRGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007; 356(2): 323-328.
- Holmes K, Roberts OL, Thomas AM, et al. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007; 19(10): 2003-2012.
- Board R, Jayson GC. Platelet-derived growth factor receptor (PDGFR): A target for anticancer therapeutics. Drug Resist Updat. 2005; 8(1-2): 75-83.
- Östman A, Heldin CH. PDGF receptors as targets in tumor treatment. In Adv. Cancer Res., George, F.V.W.; George, K., Eds. Academic Press: 2007; 97: 247-274.
- Pietras K, Sjöblom T, Rubin K, et al. PDGF receptors as cancer drug targets. Cancer Cell. 2003; 3(5): 439-443.
- Accessdata.fda.gov. Label. 2006. [Nov 2017; Sep 26, 2018]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021938s033lbl.pdf.
- Fong TA, Shawver LK, Sun L, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (FLK-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999; 59(1): 99-106.
- Scagliotti G, Govindan R. Targeting angiogenesis with multitargeted tyrosine kinase inhibitors in the treatment of Non-small Cell Lung Cancer. Oncologist. 2010; 15(5): 436-446.
- Godl K, Gruss OJ, Eickhoff J, et al. Proteomic characterization of the angiogenesis inhibitor SU6668 reveals multiple impacts on cellular kinase signaling. Cancer Res. 2005; 65(15): 6919-6926.
- Kogan M, Fischer-Smith T, Kaminsky R, et al., J. CSF-1R up-regulation is associated with response to pharmacotherapy targeting tyrosine kinase activity in AML cell lines. Anticancer Res. 2012; 32(3): 893-899.
- Jeong WJ, Mo JH, Park MW, et al. Sunitinib inhibits papillary thyroid carcinoma with RET/PTC rearrangement but not BRAF mutation. Cancer Biol Ther. 2011; 12(5): 458-465.
- Guan H, Laird AD, Blake RA, et al. Design and synthesis of aminopropyl tetrahydroindole-based indolin-2-ones as selective and potent inhibitors of Src and Yes tyrosine kinase. Bioorg Med Chem Lett. 2004; 14(1): 187-190.
- Williams R, Berndt A, Miller S, et al. Form and flexibility in phosphoinositide 3-kinases. Biochem Soc Trans. 2009; 37(Pt4): 615-626.
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nature Rev Cancer. 2008; 8(8): 592-603.
- Kammasud N, Boonyarat C, Sanphanya K, et al. 5-Substituted pyrido[2,3-d]pyrimidine, an inhibitor against three receptor tyrosine kinases. Bioorg Med Chem Lett. 2009; 19(3): 745-750.
- Luo Y, Xiao F, Qian S, et al. Synthesis and evaluation of novel 5-sulfonyl-indolin-2-ones as potent cytotoxic agents. MedChemComm. 2011; 2(11): 1054-1057.
- Sun L, Tran N, Liang C, et al. Identification of substituted 3-[(4,5,6,7-tetrahydro-1H-indol-2-yl)methylene]- 1,3-dihydroindol-2-ones as growth factor receptor inhibitors for VEGF-R2 (FLK-1/KDR), FGF-R1, and PDGF-Rβ tyrosine kinases. J Med Chem. 2000; 43(14): 2655-2663.
- Troxler T, Greenidge P, Zimmermann K, et al. Discovery of novel indolinone-based, potent, selective and brain penetrant inhibitors of LRRK2. Bioorg Med Chem Lett. 2013; 23(14): 4085-4090.
- Henise JC, Taunton J. Irreversible Nek2 kinase inhibitors with cellular activity. J Med Chem. 2011; 54(12): 4133-4146.
- Sestito S, Nesi G, Daniele S, et al. Design and synthesis of 2-oxindole based multi-targeted inhibitors of Pdk1/Akt signaling pathway for the treatment of glioblastoma multiforme. Eur J Med Chem. 2015; 105: 274-288.
- Wang HC, Jagtap AD, Chang PT, et al. Bioisosteric replacement of an acylureido moiety attached to an indolin-2-one scaffold with a malonamido or a 2/4-pyridinoylamido moiety produces a selectively potent aurora-B inhibitor. Eur J Med Chem. 2014; 84: 312-334.
- Jagtap AD, Chang PT, Liu JR, et al. Novel acylureidoindolin-2-one derivatives as dual aurora B/Flt3 inhibitors for the treatment of acute myeloid leukemia. Eur J Med Chem. 2014; 85: 268-288.
- Yang TH, Lee CI, Huang WH, et al. Synthesis and evaluation of novel 2-pyrrolidone-fused (2-oxoindolin-3-ylidene)methylpyrrole derivatives as potential multi-target tyrosine kinase receptor inhibitors. Molecules. 2017; 22.
- Yang TH, Lee CI, Huang WH, et al. Structural optimization and evaluation of novel 2-pyrrolidone-fused (2-oxoindolin-3-ylidene)methylpyrrole derivatives as potential VEGFR-2/PDGFRβ inhibitors. Chem Cent J. 2017; 11: 72.
- Ding L, Tang F, Huang W, et al. Design, synthesis, and biological evaluation of novel 3-pyrrolo[b]cyclohexylene-2-dihydroindolinone derivatives as potent receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2013; 23(20): 5630-5633.
- Li X, Huang P, Cui JJ, et al. Novel pyrrolyllactone and pyrrolyllactam indolinones as potent cyclin-dependent kinase 2 inhibitors. Bioorg Med Chem Lett. 2003; 13(11): 1939-1942.
- Karnthaler-Benbakka C, Groza D, Koblmüller B, et al. Targeting a targeted drug: an approach toward hypoxia-activatable tyrosine kinase inhibitor prodrugs. ChemMedChem. 2016; 11: 2410-2421.
- Jin YZ, Fu DX, Ma N, et al. Synthesis and biological evaluation of 3-substituted-indolin-2-one derivatives containing chloropyrrole moieties. Molecules. 2011; 16(11): 9368-9385.
- Sun L, Liang C, Shirazian S, et al. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003; 46(7): 1116-1119.
- Amaya GM, Durandis R, Bourgeois DS, et al. Cytochromes p450 1A2 and 3A4 catalyze the metabolic activation of sunitinib. Chem Res Toxicol. 2018; 31(7): 570-584.
- Harmsen S, Dolman ME, Nemes Z, et al. Development of a cell-selective and intrinsically active multikinase inhibitor bioconjugate. Bioconjugate Chem. 2011; 22(4): 540-545.
- Sartori A, Portioli E, Battistini L, et al. Synthesis of novel c(AmpRGD)–sunitinib dual conjugates as molecular tools targeting the αvβ3 integrin/VEGFR2 couple and impairing tumor-associated angiogenesis. J Med Chem. 2017; 60(1): 248-262.
- Yang T, Ng WH, Chen H, et al. Mitochondrial-targeting Met kinase inhibitor kills erlotinib-resistant lung cancer cells. ACS Med Chem Lett. 2016; 7(8): 807-812.
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015; 36(7): 422-439.
- Laufer SA, Domeyer DM, Scior TR, et al. Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. J Med Chem. 2005; 48(3): 710-722.
- Chiang CC, Lin YH, Lin SF, et al. Discovery of pyrrole-indoline-2-ones as aurora kinase inhibitors with a different inhibition profile. J Med Chem. 2010; 53(16): 5929-5941.
- Zhang L, Zheng Q, Yang Y, et al. Synthesis and in vivo SAR study of indolin-2-one-based multi-targeted inhibitors as potential anticancer agents. Eur J Med Chem. 2014; 82: 139-151.
- Ma F, Liu P, Lei M, et al. Design, synthesis and biological evaluation of indolin-2-one-based derivatives as potent, selective and efficacious inhibitors of FMS-like tyrosine kinase3 (Flt3). Eur J Med Chem. 2017; 127: 72-86.
- Cho TP, Dong SY, Jun F, et al. Novel potent orally active multitargeted receptor tyrosine kinase inhibitors: synthesis, structure-activity relationships, and antitumor activities of 2-indolinone derivatives. J Med Chem. 2010; 53(22): 8140-8149.
- Xu RH, Shen L, Wang KM, et al. Famitinib versus placebo in the treatment of refractory metastatic colorectal cancer: a multicenter, randomized, double-blinded, placebo-controlled, phase II clinical trial. Chin J Cancer. 2017; 36(1): 97.
- Informed Health Online. Nintedanib (Vargatef) for advanced lung cancer: Overview. Apr 1, 2015. [Apr 1, 2015; Nov 26, 2018] Available from: https://www.ncbi.nlm.nih.gov/books/NBK310261.
- Li X, Huang P, Cui JJ, et al. Novel pyrrolyllactone and pyrrolyllactam indolinones as potent cyclin-dependent kinase 2 inhibitors. Bioorg Med Chem Lett. 2003; 13(11): 1939-1942.
- Islam I, Bryant J, Chou YL, et al. Indolinone based phosphoinositide-dependent kinase-1 (PDK1) inhibitors. Part 1: Design, synthesis and biological activity. Bioorg Med Chem Lett. 2007; 17(14): 3814-3818.
- Buchy E, Valetti S, Mura S, et al. Synthesis and cytotoxic activity of self-assembling squalene conjugates of 3-[(pyrrol-2-yl)methylidene]-2,3-dihydro-1H-indol-2-one anticancer agents. Eur J Med Chem. 2014; 2015(1): 202-212.
- Huang Q, Zhou C, Chen X, et al. Prodrug AST-003 improves the therapeutic index of the multi-targeted tyrosine kinase inhibitor sunitinib. PloS One. 2015; 10(10): e0141395.
- Lv K, Wang LL, Liu ML, et al. Synthesis and antitumor activity of 5-[1-(3-(dimethylamino)propyl)-5-halogenated-2-oxoindolin-(3Z)-ylidenemethyl]-2,4 -dimethyl-1h-pyrrole-3-carboxamides. Bioorg Med Chem Lett. 2011; 21(10): 3062-3065.
- Sun L, Tran N, Liang C, et al. Design, synthesis, and evaluations of substituted 3-[(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases. J Med Chem. 1999; 42(25): 5120-5130.
- Jiangsu HengRui Medicine Co., Ltd. ClinicalTrials.gov. Safety and Efficacy Study of Famitinib in Patients With Advanced Colorectal Adenocarcinoma (FACT) (FACT). March 18, 2015. [Apr 17, 2018; Nov 26, 2018]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02390947.