
Development of a Companion Diagnostic PD-L1 Immunohistochemistry Assay for Pembrolizumab Therapy in Head and Neck Squamous Cell Carcinoma
Christopher J. La Placa1*, Monika D. Vilardo1, Brittany M. Watts1, Monika D. Polewski1, Siena Tabuena-Frolli1, Malinka Jansson1, Kenneth Emancipator1, Fan Jin1, Burak Gumuscu1, Joy Y. Ge2, Michael DiMaio1, Karina Kulangara1*
1Agilent Technologies, Inc. 6392 Via Real, Carpinteria, California, United States
2Merck & Co., Inc. 2000 Galloping Hill Road, Kenilworth, New Jersey, United States
Abstract
Objectives: FDA approval of PD-L1 IHC 22C3 pharmDx for use as an aid in identifying head and neck squamous cell carcinoma (HNSCC) patients for treatment with pembrolizumab was based on the results of rigorous analytical and clinical validation testing.
Methods: For the HNSCC indication, the device was validated at Agilent Technologies on the performance of sensitivity and precision using the Combined Positive Score (CPS) ≥ 1 and CPS ≥ 20 cutoffs; external validation studies were performed at three external laboratories. CPS ≥ 1 and CPS ≥ 20 cutoffs were evaluated in KEYNOTE-048, a phase 3 clinical trial.
Results: Analytical validation studies supporting the companion diagnostic indication (CPS ≥ 1) achieved point estimates of > 85% for negative, positive, and overall percent agreement. Clinical validation studies show that HNSCC patients treated with pembrolizumab as a single agent had an overall survival (OS) of 12.3 months at CPS ≥ 1 (95% CI, 10.8-14.9) compared with patients receiving cetuximab, platinum, and 5-fluorouracil (CPS ≥ 1 OS of 10.3 months (95% CI, 9.0-11.5)).
Conclusion: Analytical and clinical validation studies demonstrate that PD-L1 IHC 22C3 pharmDx is a precise companion diagnostic assay, allowing for selection of eligible HNSCC patients for treatment with pembrolizumab.
Introduction
The ability of tumor cells to bypass the immune system’s natural process of detecting and destroying malignancies is a key point of interest in the realm of immuno-oncology1. However, this process can be interrupted by a tumor’s potential to evade immune surveillance via T-cell checkpoint pathways, thereby thwarting an effective immune response and allowing the tumor to recur and/or metastasize2,3. Programmed cell death 1 (PD-1), a negative costimulatory receptor, is often expressed on the surface of activated T cells, B cells, and macrophages4,5. Its ligands, PD-L1 and PD-L2, are often expressed on the surface of tumor cells3,4, as well as on immune cells6. Binding of PD-L1 on tumor and immune cells to the PD-1 receptor found on T-cells may lead to inhibition of T-cell receptor-mediated lymphocyte proliferation and cytokine secretion, causing a decreased antitumor response and potentially contributing to poor prognosis1. An increased understanding of dysregulation and evasion of the immune system via the PD-L1 pathway has resulted in improved options for immunotherapeutic intervention, significantly benefiting cancer patients who fall into relevant clinical populations.
Preliminary biomarker studies investigating the PD-1 pathway blockade, which inhibits binding of PD-1 and its ligands, have demonstrated a correlation between pre-treatment PD-L1 expression and response to treatment with anti-PD-1 therapy4. These studies indicate that PD-L1 expression in tumor cells and/or tumor associated inflammatory cells may be predictive of improved clinical outcomes when patients are stratified by biomarker expression4,5. Adaptive immune resistance caused by the PD-1:PD-L1 interaction has been observed in head and neck squamous cell carcinoma (HNSCC) in multiple independent studies7,8. Furthermore, HNSCC encompasses a unique patient population including malignancies of the oral cavity, oropharynx, hypopharynx, and larynx, providing a rationale for developing a therapeutic and a reliable accompanying device to address an unmet medical need in the clinical oncology space.
Pembrolizumab, an anti-PD-1 immunoglobulin G4 kappa isotype antibody developed by Merck & Co, Inc., Kenilworth, NJ, USA, has demonstrated efficacy in multiple clinical trials9-12, including a phase III clinical trial (KEYNOTE-048; NCT02358031) for patients with recurrent or metastatic HNSCC9,12. To determine which patients can be safely and effectively treated with pembrolizumab, a precise and robust biomarker assay, PD-L1 IHC 22C3 pharmDx, was utilized9.
PD-L1 immunohistochemistry (IHC) assays have been developed for HNSCC13; however, none have previously been cleared or approved as companion diagnostic devices by the FDA14. Each one of these IHC assays is associated with a unique scoring approach, as well as a different primary antibody clone and isotype, secondary antibody/detection reagent formulations, and IHC protocols11. PD-L1 IHC 22C3 pharmDx, an IHC assay using the monoclonal mouse anti-PD-L1, clone 22C3, antibody has been developed and was utilized to determine PD-L1 expression in a phase III clinical trial (KEYNOTE-048) for patients with recurrent or metastatic HNSCC. The trial demonstrated significant improvement in overall survival (OS) for the subgroup of patients with PD-L1 Combined Positive Score (CPS) ≥ 1 randomized to pembrolizumab as a single agent compared to those randomized to cetuximab in combination with chemotherapy9,12 (CPS is discussed below in the Scoring Interpretation section). This paper presents the analytical and clinical validation results for PD-L1 IHC 22C3 pharmDx with respect to the HNSCC indication.
Materials and Methods
PD-L1 IHC 22C3 pharmDx was analytically validated on the performance of parameters including sensitivity, precision, and reproducibility. Internal analytical validation studies performed at Agilent Technologies included sample sizes ranging from 24 to 48 specimens with multiple replicates or reads per replicate, depending on the study and its design. External validation studies (performed at three CAP accredited and/or CLIA licensed laboratories) included sample sizes ranging from 38 to 62 specimens, depending on the specific study.
Tissue Specimen Preparation
For the analytical validation, all specimens tested were formalin-fixed, paraffin-embedded (FFPE) archival human tissue, and were not collected from patients enrolled in KEYNOTE-048. Tissues were sectioned at 4µm thickness, placed on positively charged glass slides, and oven-dried at 58 ±2 °C for approximately 1 hour. Prepared slides were then stored in the dark at 2-8 °C and stained using PD-L1 IHC 22C3 pharmDx within 6 months of microtomy.
Staining Procedure
A 3-in-1 procedure encompassing deparaffinization, rehydration, and target retrieval was performed using EnVision FLEX Target Retrieval Solution, Low pH (Agilent code K8005) to pre-treat all specimens in the PT Link. The Autostainer Link 48, an automated IHC testing platform with a staining protocol validated for PD-L1 IHC 22C3 pharmDx (Agilent code SK006), was used to perform all IHC testing with EnVision FLEX Wash Buffer (Agilent code K8007). PD-L1 IHC 22C3 pharmDx contains optimized reagents and the protocol required to complete an IHC testing procedure of FFPE specimens using Autostainer Link 48. Following incubation with the primary monoclonal antibody to PD-L1 or the Negative Control Reagent, specimens were incubated with a Linker antibody specific to the host species of the primary antibody, and then were incubated with a ready-to-use visualization reagent consisting of secondary antibody molecules and horseradish peroxidase molecules coupled to a dextran polymer backbone. The enzymatic conversion of the subsequently added chromogen, 3,3’-diaminobenzidine tetrahydrochloride, results in precipitation of a visible reaction product at the site of the antigen. The color of the chromogenic reaction is modified by a chromogen enhancement reagent. The stained specimens were counterstained with hematoxylin (Agilent code K8008) and coverslipped. All reagents and instrumentation were provided by Agilent Technologies.
Scoring
For determination of PD-L1 expression level, the CPS algorithm was used, with specific focus on two clinical diagnostic cutoffs: CPS ≥ 1 and CPS ≥ 20. The scoring algorithm is defined per the equation below and Table 1:
Prior to scoring for the analytical validation studies and clinical trial, a training effectiveness procedure was performed requiring observers to score 30 unique HNSCC cases stained with PD-L1 IHC 22C3 pharmDx at the CPS ≥ 1 and CPS ≥ 20 cutoffs. Prior to participation in the studies, observers were expected to demonstrate ≥85% inter-observer overall percent agreement (OA) and ≥85% intra-observer OA, based on positive and negative diagnostic outcome for each cutoff.
Table 1: CPS Numerator Inclusion/Exclusion Criteria for HNSCC
|
Tissue Elements |
Included in the Numerator |
Excluded from the Numerator |
|
Tumor Cells |
· Convincing partial or complete linear membrane staining (at any intensity) of viable invasive tumor cells |
· Non-staining tumor cells · Tumor cells with only cytoplasmic staining
|
|
Immune Cells |
· Membrane and/or cytoplasmica staining (at any intensity) of mononuclear inflammatory cells (MICs) within tumor nests and adjacent supporting stromab o Lymphocytes (including lymphocyte aggregates) o Macrophagesc
· Only MICs directly associated with the response to the tumor are scored. |
· Non-staining MICs · MICs (including lymphoid aggregates) associated with ulcers or other inflammatory processes · MICs associated with carcinoma in situ · MICs associated with benign structures · Neutrophils, eosinophils and plasma cells |
|
Other Cells |
· Not included |
· Carcinoma in situ · Benign cells · Stromal cells (including fibroblasts) · Necrotic cells and/or cellular debris |
aIn MICs, membrane and cytoplasmic staining are often indistinguishable due to high nuclear to cytoplasmic ratio. Therefore, membrane and/or cytoplasmic staining of MICs is included in the score.
bAdjacent MICs are defined as being within the same 20x field as the tumor. However, MICs that are NOT directly associated with the response to the tumor should be excluded.
cMacrophages and histiocytes are considered the same cells.
Phase 3 Clinical Trial
The efficacy of pembrolizumab (KEYTRUDA) was investigated in KEYNOTE-048 (NCT02358031), a randomized, multicenter, open label, active controlled trial conducted in 882 patients with metastatic or recurrent HNSCC, who had not previously received systemic therapy for metastatic disease or with recurrent disease who were considered incurable by local therapies12. Patients were randomized 1:1:1 to one of the following treatment arms:
- KEYTRUDA 200 mg intravenously every 3 weeks
- KEYTRUDA 200 mg intravenously every 3 weeks, carboplatin AUC 5 mg/mL/min intravenously every 3 weeks or cisplatin 100 mg/m2 intravenously every 3 weeks, and fluorouracil (FU) 1000 mg/m2/day as a continuous intravenous infusion over 96 hours every 3 weeks (maximum of 6 cycles of platinum and FU)
- Cetuximab 400 mg/m2 intravenously as the initial dose then 250 mg/m2 intravenously once weekly, carboplatin AUC 5 mg/mL/min intravenously every 3 weeks or cisplatin 100 mg/m2 intravenously every 3 weeks, and FU 1000 mg/m2/day as a continuous intravenous infusion over 96 hours every 3 weeks (maximum of 6 cycles of platinum and FU)
The main efficacy outcome measures were OS and progression free survival (PFS) as assessed by blinded independent central review (BICR) according to response evaluation criteria in solid tumors (RECIST) v1.1 (modified to follow a maximum of 10 target lesions and a maximum of five target lesions per organ) sequentially tested in the subgroup of patients with CPS ≥ 20, the subgroup of patients with CPS ≥ 1, and the overall population12.
Statistical Analysis (Analytical Validation)
Comparisons between the IHC status (positive/negative) of each test condition and the consensus (most frequently occurring diagnostic observation) were made for each specimen. Comparison results were subsequently pooled across specimens for calculations of percent agreements. Negative percent agreement (NPA), positive percent agreement (PPA), and OA were calculated for each precision and reproducibility study, with corresponding two-sided 95% percentile bootstrap confidence intervals (CIs). Since the percentile bootstrap method cannot compute CIs when 100% agreement is observed, Wilson score confidence intervals were reported for studies resulting in zero discordant comparisons.
The acceptance criteria were defined as: the lower bound value of the two-sided 95% CIs computed on percent agreement must meet or exceed 85%. The data presented herein were analyzed using NPA, PPA, and OA.
Results
Internal Validation
Sensitivity
The prevalence of PD-L1 protein in FFPE HNSCC human tissue specimens was assessed using PD-L1 IHC 22C3 pharmDx. One hundred twelve unique archival HNSCC specimens, represented as whole tissue specimens, including primary tumors and metastases, early (Stage I-II) and late (Stage III-IV) stage disease, were tested. These specimens were evaluated based on CPS (see Scoring Interpretation section).
PD-L1 expression was observed across the dynamic range from CPS 0 to CPS 100 (Figures 1, 2). The ability of PD-L1 IHC 22C3 pharmDx to detect the target protein across a dynamic range of expression (including primary and metastatic tumors and early and late stage disease) ensures that the device can be used for patients whose tumors express PD-L1 at all levels.
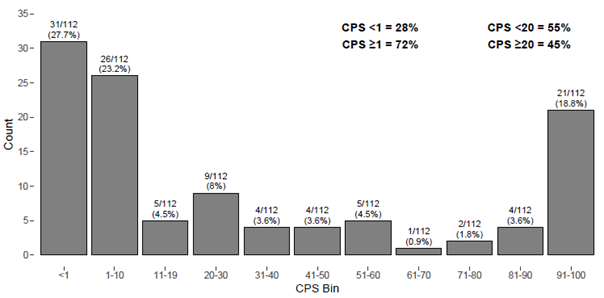
Figure 1: Analytical sensitivity of PD-L1 IHC 22C3 pharmDx tested on HNSCC specimens using the CPS ≥ 1 and CPS ≥ 20 cutoffs. Using the CPS ≥ 1 cutoff, 28% of specimens had no PD-L1 expression (CPS < 1) and 72% of specimens had PD-L1 expression (CPS ≥ 1). Using the CPS ≥ 20 cutoff, 55% of specimens had no PD-L1 expression (CPS < 20) and 45% of specimens had PD-L1 expression (CPS ≥ 20).
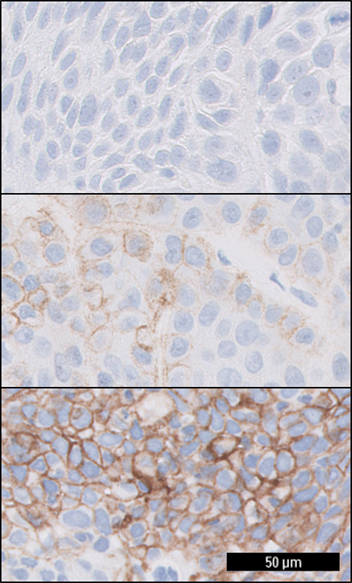
Figure 2: HNSCC tissue specimens stained with PD-L1 IHC 22C3 pharmDx. The upper image depicts HNSCC tissue with a CPS of 0, the middle image with a CPS of 40, and the lower image with a CPS of 95.
Precision
Assay precision was measured on the basis of inter-/intra-observer precision, combined precision (inter-instrument/operator/day/lot), and intra-run repeatability at Agilent Technologies using the CPS ≥ 1 and CPS ≥ 20 cutoffs as part of internal analytical validation.
All studies were analyzed based on diagnostic agreement (above or below cutoff) between replicates of specimens, with respect to the CPS ≥ 1 and CPS ≥ 20 cutoffs. The two cutoffs were evaluated in separate analytical validation studies and scored independently from one another. For the CPS ≥ 1 cutoff, PD-L1 expression levels are defined as CPS < 1 and CPS ≥ 1. For the CPS ≥ 20 cutoff, PD-L1 expression levels are defined as CPS < 20 and CPS ≥ 20.
Scoring precision was evaluated on the performance of inter-/intra-observer precision for the CPS ≥ 1 and CPS ≥ 20 cutoffs, independently. Three trained and certified pathologists (observers) reviewed a set of stained slides three times, with evaluation order re-randomized between reads. A minimum washout period of 14 days was implemented between each read to minimize recall bias. Diagnostic agreement was evaluated among the three observers over the three reads to determine inter-observer precision. For CPS ≥ 1, point estimates for NPA, PPA, and OA of 88.9%, 99.1%, and 94.4% were achieved, with lower bound values of 78.8%, 97.4%, and 89.8%, respectively. For CPS ≥ 20, point estimates for NPA, PPA, and OA of 96.3%, 93.1%, and 94.9% were achieved, with lower bound values of 91.8%, 87.3%, and 91.4%, respectively (Figure 3).
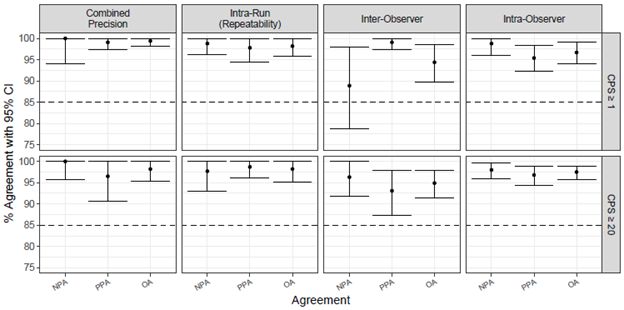
Figure 3: Summary of percent agreement for PD-L1 IHC 22C3 pharmDx precision studies performed at Agilent Technologies. For CPS ≥ 1, all study parameters other than inter-observer NPA met acceptance criteria. For CPS ≥ 20, all study parameters met acceptance criteria.
Analysis of diagnostic agreement in the CPS ≥ 1 internal inter-observer precision study resulted in 214 comparisons to consensus, with 202 concordant (88 concordant negative, 114 concordant positive) and 12 discordant comparisons (1 discordant negative, 11 discordant positive). The sample size (N=24) in combination with a high enrichment of challenging cases (58.3% near cutoff) contributed to the failure to meet acceptance criteria for NPA. A larger sample size (N=62) was included in the external inter-observer reproducibility study at CPS ≥ 1, resulting in passing results per the prespecified acceptance criteria (see Observer Precision section).
For the intra-observer precision study at CPS ≥ 1, point estimates for NPA, PPA, and OA of 98.8%, 95.4%, and 96.7% were achieved, respectively, with lower bound values of 96.0%, 92.3%, and 94.0%, respectively. For intra-observer precision at CPS ≥ 20, point estimates for NPA, PPA, and OA of 98.0%, 96.8%, and 97.5% were achieved, with lower bound values of 95.9%, 94.4%, and 95.8%, respectively (Figure 3). These results demonstrate that HNSCC specimens can be assigned a consistent diagnostic status by the same observer at the CPS ≥ 1 and CPS ≥ 20 cutoffs, confirming observer precision of the CPS scoring algorithm at the respective cutoffs for this indication.
Intermediate precision (also known as combined precision) was evaluated by investigating the combined effect of inter-operator, inter-instrument, inter-day, and inter-lot variables. All endpoints (NPA, PPA and OA) for the combined precision study met acceptance criteria at both CPS ≥ 1 and CPS ≥ 20 cutoffs. At the CPS ≥ 1 cutoff, point estimates for NPA, PPA, and OA of 100%, 99.1% and 99.4% were achieved, with lower bound values of 94.0%, 97.3%, and 98.2%, respectively. At the CPS ≥ 20 cutoff, point estimates for NPA, PPA, and OA of 100%, 96.5% and 98.2% were achieved, with lower bound values of 95.7%, 90.6%, and 95.3%, respectively (Figure 3).
Additionally, intra-run repeatability was evaluated. Six serial sections from each specimen were tested using PD-L1 IHC 22C3 pharmDx on one instrument simultaneously; five sections were stained with the primary antibody, and one section was stained with the Negative Control Reagent. For the intra-run repeatability at CPS ≥ 1, point estimates for NPA, PPA, and OA of 98.8%, 97.8%, and 98.2% were achieved, with lower bound values of 96.2%, 94.4%, and 95.9%, respectively. For the intra-run repeatability at CPS ≥ 20, point estimates for NPA, PPA, and OA of 97.7%, 98.7%, and 98.2% were achieved, with lower bound values of 92.9%, 96.2%, 95.2%, respectively (Figure 3).
The internal analytical validation studies demonstrate that PD-L1 IHC 22C3 pharmDx is a precise and repeatable assay at the CPS ≥ 1 and CPS ≥ 20 cutoffs on HNSCC specimens. Although all endpoints were not met for the internal inter-observer precision study, results from an additional, more stringent study (see Observer Precision section) suggest that HNSCC specimens stained with PD-L1 IHC 22C3 pharmDx can be reliably and reproducibly scored by multiple pathologists and in multiple reads at both the CPS ≥ 1 and CPS ≥ 20 cutoffs.
External Validation
Inter- and intra-site reproducibility was conducted at three external CAP accredited and/or CLIA licensed laboratories evaluating at both the CPS ≥ 1 and CPS ≥ 20 cutoffs. One operator from each laboratory performed five automated IHC testing runs using PD-L1 IHC 22C3 pharmDx over 5 non-consecutive days. Each staining run contained replicate sections from the same set of HNSCC specimens (n=38), with one slide stained using the NCR and one slide stained with the anti- PD-L1 (clone 22C3) primary antibody. Efforts were made to balance the proportion of positive and negative specimens at each respective cutoff, as well as to include approximately 25% of near cutoff specimens, considered to be challenging cases (CPS <1 – CPS 10 for the CPS ≥ 1 cutoff; CPS 10 – CPS 30 for the CPS ≥ 20 cutoff). Staining was evaluated at the CPS ≥ 1 and CPS ≥ 20 cutoffs by a single pathologist at each of the three external laboratories, with a minimum washout period of 14 days between each read to minimize recall bias.
Inter-/intra-observer precision was assessed through blinded and randomized slide evaluation at the three external sites at both cutoffs. One trained and certified pathologist at each site performed three independent evaluations of a stained set of 62 HNSCC slides representing a dynamic range of PD-L1 expression; PD-L1-positive, negative, and near cutoff specimens with respect to each cutoff were included in the set. Unique “wildcard” slides were included in the evaluation but were excluded from the analyzed dataset. A minimum washout period of 14 days between each read was implemented to minimize recall bias.
Assay Reproducibility
For the inter-site reproducibility at CPS ≥ 1, point estimates for NPA, PPA, and OA of 96.8%, 93.3%, and 95.1% were achieved, with lower bound values of 92.6%, 86.7%, and 91.2%, respectively. For the inter-site reproducibility at CPS ≥ 20, point estimates for NPA, PPA, and OA of 95.5%, 81.0%, and 90.5% were achieved, with lower bound values of 92.0%, 71.3%, and 86.5%, respectively (Figure 4). Inter-site reproducibility PPA at CPS ≥ 20 did not meet acceptance criteria. Many of the variables from inter-site reproducibility were also evaluated internally in combined precision, which measured the compounded effects of inter-instrument/-operator/-day/-lot, and met all acceptance criteria (see Assay Precision section).
Intra-site reproducibility was evaluated by testing the reproducibility within a site, across each of the five testing runs. For intra-site reproducibility at CPS ≥ 1, point estimates for NPA, PPA, and OA of 95.7%, 97.0%, and 96.3% were achieved, with lower bound values of 91.3%, 94.5%, and 93.5%, respectively. For intra-site reproducibility at CPS ≥ 20, point estimates for NPA, PPA, and OA of 96.9%, 90.6%, and 94.9% were achieved, with lower bound values of 94.6%, 86.3%, and 92.8%, respectively (Figure 4). Because all intra-site reproducibility study parameters met acceptance criteria, this study demonstrates that PD-L1 IHC 22C3 pharmDx is reproducible within the same site over multiple days and runs.
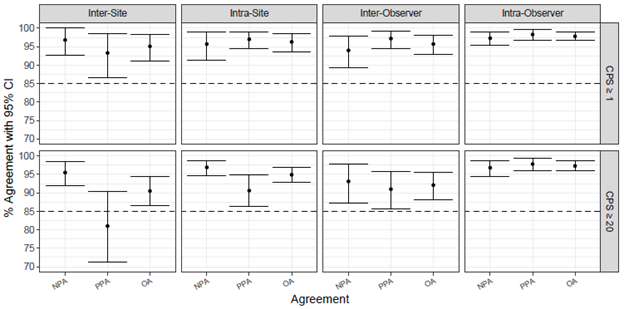
Figure 4: Summary of percent agreement for PD-L1 IHC 22C3 pharmDx precision studies performed at three external sites. For CPS ≥ 1, all study parameters met acceptance criteria. For CPS ≥ 20, all study parameters other than inter-site PPA met acceptance criteria.
Observer Precision
Inter-observer precision was evaluated externally by testing scoring reproducibility between three pathologists using 62 HNSCC specimens stained with PD-L1 IHC 22C3 pharmDx. At CPS ≥ 1, point estimates for NPA, PPA, and OA of 94.0%, 97.2%, and 95.7% were achieved, with lower bound values of 89.3%, 94.4%, and 93.0%, respectively. At CPS ≥ 20, point estimates for NPA, PPA, and OA of 93.1%, 91.0%, and 92.1% were achieved, with lower bound values of 87.2%, 85.7%, and 88.2%, respectively. Intra-observer precision was evaluated by testing scoring reproducibility within each of the three external pathologists using the same set of scores from the 62 specimens over three blinded and randomized reads. At CPS ≥ 1, point estimates for NPA, PPA, and OA of 97.3%, 98.3%, and 97.8% were achieved, with lower bound values of 95.4%, 96.8%, and 96.8%, respectively. At CPS ≥ 20, point estimates for NPA, PPA, and OA of 96.8%, 97.8%, and 97.3% were achieved, with lower bound values of 94.5%, 96.0%, and 95.9%, respectively (Figure 4). These results demonstrate that the CPS ≥ 1 and CPS ≥ 20 cutoffs, when used on HNSCC specimens stained with PD-L1 IHC 22C3 pharmDx, are reproducible within and between observers.
Clinical Validation
KEYNOTE-048
A total of 601 patients were randomized to KEYTRUDA as a single agent or cetuximab in combination with chemotherapy arms; 301 patients to KEYTRUDA as a single agent arm and 300 patients to the cetuximab in combination with chemotherapy arm12. The study population characteristics were: median age of 61 years (range: 22 to 94); 36% age 65 or older; 85% male; 74% White and 19% Asian, and 1.7% Black; 61% ECOG PS of 1; and 79% were former/current smokers12.
For the subgroup of patients randomized to KEYTRUDA as a single agent or to cetuximab in combination with chemotherapy arms, PD-L1 expression level was determined using PD-L1 IHC 22C3 pharmDx. Overall, 85% (512/601) of the patients had tumors that expressed PD-L1 at CPS ≥ 112. Eighty-six percent (380/442) of patients whose tumors were newly obtained for PD-L1 testing and 83% (132/159) of patients whose archival tumors were tested expressed PD-L1 at CPS ≥ 1. Forty-three percent (255/597) of the patients had tumors that expressed PD-L1 with CPS ≥ 20; four patients had unknown PD-L1 expression status (one specimen was archival tissue and three specimens were newly obtained tissue). Forty-two percent (186/439) of patients whose tumors were newly obtained for PD-L1 testing and 44% (69/158) of patients whose archival tumors were tested expressed PD-L1 at CPS ≥ 2012.
The treatment effect of pembrolizumab in patients expressing PD-L1 at CPS ≥ 1 and CPS ≥ 20 was both statistically significant and clinically meaningful, with increasing efficacy correlated with increasing PD-L1 expression12. Pembrolizumab as a single agent yielded a median OS of 12.3 months at CPS ≥ 1 (95% CI, 10.8-14.9) and 14.9 months at CPS ≥ 20 (95% CI, 11.6-21.5), compared with patients receiving cetuximab, platinum, and fluorouracil, which had a median OS of 10.3 months at CPS ≥ 1 (95% CI, 9.0-11.5) and 10.7 months at CPS ≥ 20 (95% CI, 8.8-12.8) 12. In an exploratory subgroup analysis conducted using data from patients with CPS 1-19, pembrolizumab as a single agent yielded a median OS of 10.8 months (95% CI, 9.0-12.6), compared with patients receiving cetuximab, platinum, and fluorouracil, which had a median OS of 10.1 months (95% CI, 8.7-12.1)15. For comparison, in the total study population (irrespective of biomarker cutoff categories) pembrolizumab as a single agent demonstrated non-inferiority, but not superiority compared to cetuximab, platinum, and fluorouracil treatment. Specifically, the median OS was 11.6 months (95% CI, 10.5-13.6), compared with patients receiving cetuximab, platinum, and fluorouracil, which had a median OS of 10.7 months (95% CI, 9.3-11.7)12. These clinical results demonstrate that pembrolizumab therapy is an effective treatment option and may contribute to an increased lifespan for patients with HNSCC in both biomarker cutoff categories (CPS ≥ 1 and CPS ≥ 20).
Discussion
PD-L1 IHC 22C3 pharmDx was codeveloped with pembrolizumab in order to determine HNSCC patient eligibility for pembrolizumab. The performance of PD-L1 IHC 22C3 pharmDx was supported by analytical validation studies described above, with acceptable or justifiable results for all parameters investigated at both the CPS ≥ 1 and CPS ≥ 20 cutoffs. Pre-determined acceptance criteria were met for all statistical endpoints (NPA, PPA, and OA) for both cutoffs, with the exception of CPS ≥ 1 internal inter-observer NPA and CPS ≥ 20 inter-site PPA.
PD-L1 IHC 22C3 pharmDx was analytically validated yielding precise and reliable results at CPS ≥ 1, the cutoff used to determine eligibility for KEYTRUDA. Some factors with the potential to introduce diagnostic variability in a clinical setting are isolated in the precision and reproducibility portions of the analytical validation studies; these include variables within/between laboratories (inter-instrument/-operator/-day/-lot, intra-run) as well as variables within/between pathologists (and/or observers). Performing external analytical validation studies in addition to internal analytical validation studies provides a more robust understanding of the performance of the diagnostic assay under use of the end-user laboratory. For CPS ≥ 1, the cutoff used to determine eligibility for KEYTRUDA, all analytical studies produced acceptable results with the exception of the internal inter-observer study, which did not meet acceptance criteria for NPA due to potential contributing factors such as a limited sample size and a high enrichment of challenging cases (58.3% near cutoff). However, the subsequent external inter-observer reproducibility study (which had a greater sample size) did meet acceptance criteria. At the CPS ≥ 20 cutoff, the external reproducibility study did not meet acceptance criteria for inter-site PPA, but did meet all acceptance criteria for intra-site, inter-observer, and intra-observer reproducibility, as well as inter-site reproducibility NPA and OA.
The clinical validation results demonstrated superior OS for HNSCC patients expressing CPS ≥ 1 when treated with KEYTRUDA as a single agent, as compared to patients receiving cetuximab, platinum, and fluorouracil12. PD-L1 IHC 22C3 pharmDx is a sensitive and precise companion diagnostic assay, providing clinical utility in determining HNSCC patient eligibility for pembrolizumab therapy using the CPS ≥ 1 cutoff. Similarly, superior OS for HNSCC patients expressing CPS ≥ 20 when treated with KEYTRUDA as a single agent was observed, as compared to patients receiving cetuximab, platinum, and fluorouracil12. CPS ≥ 1, the cutoff with the potential to reach the largest population of patients who may benefit from treatment with KEYTRUDA monotherapy, was selected as the therapeutic and companion diagnostic cutoff, while the clinical outcomes for patients in the CPS ≥ 20 subgroup are also included in the product labelling. Relative to standard of care therapy, both cutoffs (CPS ≥ 1, CPS ≥ 20) for KEYTRUDA monotherapy are associated with a significant survival benefit12.
FDA approval of PD-L1 IHC 22C3 pharmDx as a companion diagnostic device fulfills an unmet medical need for an array of patients who fall into the KEYNOTE-048 population, which includes malignancies of the oral cavity, oropharynx, hypopharynx, and larynx. The FDA approval of PD-L1 IHC 22C3 pharmDx as a companion diagnostic device to aid in identifying HNSCC patients for treatment with pembrolizumab was preceded by approvals for non-small cell lung cancer, gastric or gastroesophageal junction adenocarcinoma, cervical cancer, and urothelial carcinoma, and was followed by approvals for esophageal squamous cell carcinoma and triple negative breast cancer. For all approved tumor indications, precision of the diagnostic assay was confirmed in analytical validation studies, and clinical performance was evaluated in clinical trials. While the specific populations of eligible patients and associated treatment regimens vary by tumor indication, PD-L1 IHC 22C3 pharmDx is FDA-approved for use as a companion diagnostic to KEYTRUDA in each of the approved tumor indications listed above. Though other anti-PD-L1 primary antibodies exist, PD-L1 IHC 22C3 pharmDx is the only diagnostic assay approved by FDA for determination of HNSCC patient eligibility for KEYTRUDA.
Acknowledgements
This work was supported by Merck & Co., Inc; all analytical and clinical validation studies presented in this manuscript were made possible by their sponsorship.
Conflict of Interest Statement
Authors denoted with “a” are current or former employees of Agilent Technologies, Inc; authors denoted with “b” are current or former employees of Merck & Co., Inc. All analytical and clinical studies presented in this manuscript were funded by Merck & Co., Inc.
Ethics Statement
The ASTERAND Human Tissue Biorepository upholds the fundamental bioethics principles of autonomy, privacy, safety and informed consent. We support legal and ethical efforts to promote voluntary donation of surplus human tissues and clinical information for research use.
BioIVT maintains a global network of collaborators who conduct subject recruitment, informed consent and scientific activities. We ensure that all of our activities, and those of our collaborators, are conducted in accordance with applicable laws, regulations, and ordinances. BioIVT confirms that the following activities have been completed by our collaborators (as necessary): institutional and independent review board approval, privacy officer authorization, government licenses, or industry accreditations. We continually monitor our practices to conform to evolving external scientific, legal and ethical requirements.
References
- Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-1034. https://dx.doi.org/10.1084%2Fjem.192.7.1027
- Ferris R. Immunology and Immunotherapy of Head and Neck Cancer. J Clin Oncol. 2015;33:3293-3304. https://dx.doi.org/10.1200%2FJCO.2015.61.1509
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674. https://doi.org/10.1016/j.cell.2011.02.013
- Taube J, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti-PD-1 Therapy. Clin Cancer Res. 2014;20:5064-5074. https://doi.org/10.1158/1078-0432.ccr-13-3271
- Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. 2014;515:558-562. https://doi.org/10.1038/nature13904
- Kulangara K, Zhang N, Corigliano E, et al. Clinical Utility of the Combined Positive Score for Programmed Death Ligand-1 Expression and the Approval of Pembrolizumab for Treatment of Gastric Cancer. Arch Path Lab Med. 2019;143:330-337. https://doi.org/10.5858/arpa.2018-0043-oa
- Lyford-Pike S, Peng S, Young GD, et al. Evidence for a Role of the PD-1:PD-L1 Pathway in Immune Resistance of HPV-Associated Head and Neck Squamous Cell Carcinoma. Cancer Research. 2013;73:1733-1741. https://doi.org/10.1158/0008-5472.can-12-2384
- Zandberg D, Strome SE. The role of the PD-L1:PD-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncology. 2014;50:627-632. https://doi.org/10.1016/j.oraloncology.2014.04.003
- Burtness B, Harrington K, Greil R, et al. KEYNOTE-048: Phase 3 study of first-line pembrolizumab (P) for recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC). ESMO 2018 Congress. 22 October 2018.
- Chow LQM, Haddad R, Gupta S, et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma. J Clin Oncol. 2016;34:3838-3845. https://doi.org/10.1200/jco.2016.68.1478
- Roach C, Zhang N, Corigliano E, et al. Development of a Companion Diagnostic PD-L1 Immunohistochemistry Assay for Pembrolizumab Therapy in Non-Small-cell Lung Cancer. Appl Immunohistochem Mol Morphol 2016;24:392-397. https://doi.org/10.1097/pai.0000000000000408
- Burntness B, Harrington K, Greil R, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomized, open-label phase 3 study. The Lancet 2019;394(10212):1915-1928. https://doi.org/10.1016/s0140-6736(19)32591-7
- Ferris RL, Blumenschein G, Fayette J, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856-1867. https://doi.org/10.1056/nejmoa1602252
- S. Food & Drug Administration. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Retrieved on October 29th, 2019 from https://www.fda.gov/medical-devices/vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-vitro-and-imaging-tools.
- Burtness, B., Rischin, D., Greil, R., Soulières, D., Tahara, M., de Castro, G., ... & Hughes12, B. G. M. Efficacy of First-Line Pembrolizumab by PD-L1 Combined Positive Score< 1, 1-19, and≥ 20 in Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: KEYNOTE-048 Subgroup Analysis.