
Epigenetic Modifications in Ovarian Cancer: A Review
A. H. M. Zuberi Ashraf1,2, Syeda H. Afroze3, Grace A. Osuji2, Saba Y. Kayani3, Natalie Colon3, Ahmed F. Pantho3, Thomas J. Kuehl3, Kimberly A Pilkinton4, M. Nasir Uddin3,5
1Department of Science & Mathematics, Texas A&M University-Central Texas, Killeen, TX, U.S.A.
2Baylor Scott & White Health, Temple, TX, U.S.A.
3Orion Institute for Translational Medicine, Temple, TX, U.S.A.
4Department of Clinical Sciences, University of Houston College of Medicine, Houston, TX, U.S.A.
5Department of Medical Physiology, Texas A&M University College of Medicine, Temple, TX, U.S.A.
Abstract
The purpose of this article is to review the role of different epigenetic modifications in ovarian cancer. Epigenetic changes can lead to disease development, malignant transformation, and drug resistance of ovarian cancer. Silencing, methylation, and histone modification of genes contribute to ovarian cancer formation. miRNAs have frequently been found to be dysregulated in ovarian cancer cells. Cancer stem cells possess incredible DNA-repair mechanisms and higher rates of mutation; therefore, making them very invasive and resistant to most chemotherapeutic agents. The pathogenesis of ovarian cancer, types of epigenetic modifications, role of miRNA, and cancer stem cells are discussed, as well as targeting of epigenetic pathways with alternative interventions, and application of combination therapies. Using newly discovered combination therapies, it might be possible to create means to manipulate the detrimental epigenetic pathways which can lead to earlier detection, prevention, and treatment of ovarian cancer.
Ovarian cancer is responsible for approximately 1.3% of all new cancer cases but ranks fifth for cancer mortalities for women in the world1. Data from 2009-2013 suggest 12 new cases of ovarian cancer per 100,000 women per year. In addition, an estimated 22,440 new cases and 14,080 deaths from ovarian cancer occurred in 20171. There is significant patient variation in terms of disease condition and tumor types regarding ovarian cancer. Ovarian cancer is most often discovered at advanced stages as a result of limited screening procedures that are available. Ovarian tumor cell types originate from three broad categories of ovarian cells, which include surface epithelial cells, germ cells, and sex cord-stromal cells2. Table 1 reviews the different ovarian cancer cell types. Surface epithelial tumors account for approximately 85-90% of all ovarian cancer cases3. Epithelial ovarian cancer (EOC) arises from the epithelium of Müllerian structures including the ovarian surface and tubal epithelium. EOCs fall into two main histological subgroups: Type I include the slower growing low-grade serous, mucinous, endometrioid, and clear cell carcinomas3. Type II tumors are high-grade malignancies which lead to rapidly progressive disease. High-grade serous ovarian carcinoma is highly aggressive and metastatic and the most common type II tumor3.
Women with familial breast cancer type susceptibility gene (BRCA1 and BRCA2) mutations carry up to 40% elevated risk of developing ovarian cancer4. BRCA1 and BRCA2 are well-known human tumor-suppressor genes. They are both poorly conserved except in specific domains such as the BRCA1 C-terminal (BRCT) and N-terminal RING domains in BRCA1 and the BRC repeats in BRCA2. BRCA1 is universally expressed in humans, with highest levels of transcription in ovaries, testis, and thymus. BRCA2 is expressed only in actively dividing cells4. These genes are essential for maintenance of genomic stability, ensuring error-proof repair of double-strand breaks and homologous recombination2,4. Family history of ovarian, breast, and colorectal cancer in a first-degree relative could be associated with mutations in other tumor-suppressor genes such as those involved in DNA-repair pathways4. Gynecological and obstetric history of low parity, late age of first live birth, late menopause, and greater number of ovulatory cycles are also considered risk factors4. Many ovarian cancer cases are diagnosed in women aged 55-64 years, suggesting age to be the most common factor paired with other risk factors1.
The lack of effective screening methods has made ovarian cancer one of the deadliest gynecological maladies3,5. Unlike pap smears for cervical cancer or colonoscopy for colorectal cancer, ovarian cancer does not have any standardized screening procedure5. Transvaginal ultrasound and measuring levels of serum cancer antigen 125 (CA125) have been used as screening tools in randomized trials testing for ovarian cancer. CA125 is a known biomarker for tumors that is expressed at very high concentrations in ovarian cancer cells compared other types of cancers. CA125 is mainly specific to the epithelial ovarian tumors and is not necessarily specific enough for other ovarian tumors5. Further review and analysis of the data from the ovarian cancer testing trials indicate that the screening methods used did not contribute to diagnosing ovarian cancer at earlier stages or reducing overall ovarian cancer mortality. In addition, screening resulted in unnecessary surgery, psychological stress, and did not improve quality of life5. Improved general screening should involve counseling of each individual patient based on personal risk factors1,4. When ovarian cancer is diagnosed at an early stage, the 5-year survival rate is 92%, but, unfortunately, only around 15% of cases are found at this stage1,4.
This article reviews the role of some epigenetic modifications in ovarian cancer. The pathogenesis of ovarian cancer, types of epigenetic modifications in signaling pathways, DNA hyper/hypomethylation, histone modifications, enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) and polycomb repressive complexes, switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex, microRNAs, cancer stem cells (CSCs), and combination and epigenetic therapies are reviewed. In this review article, we compiled updated information from various scientific studies that investigated the molecular mechanisms in the pathogenesis of ovarian cancer. Clear and comprehensive understanding of the various molecular pathways involved in ovarian cancer formation will inform extensive research studies with a goal to establish improved methods for early disease detection and the development of advanced treatment options, resulting in more favorable prognoses for women diagnosed with ovarian cancer.
Pathogenesis of Ovarian Cancer
Comprehending the mechanisms that contribute to the development of ovarian cancer can be incredibly difficult, since early detection methods to diagnose ovarian cancer remains a challenge6. For the past three decades, there have been many studies investigating the underlying events that results in the formation of ovarian carcinoma7,8. Diversified morphology, molecular heterogeneity, and varying histological profiles of EOC are major hurdles for diagnosis during early stages of disease9. To date, several research studies were dedicated to unravelling the molecular and histological complexities of neoplastic cells that form genetically and highly unstable tumors that ultimately result in ovarian cancer10. According to recent studies, the possible sources of progenitor cells or neoplasms for ovarian cancer is not just limited to the ovaries9,11. Ovarian neoplasms are mostly of epithelial origin and fewer neoplasms mature from cell types such as germ cell, sex-cord stromal, or mixed cell types (Table 1)8. Tumors arising from the above cell types are further classified into two different types based on their distinct morphological and molecular structures. Type I tumors harbor inactivating mutations in phosphatase and tensin homolog (PTEN), switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex, and AT-rich interaction domain 1A (ARID1A), activation of the WNT-catenin, and phosphoinositide 3-kinase (PI3K) pathways (Table 2)10. Type I tumors can be further subdivided by cell of origin and histological subtype, including serous, endometrioid, clear-cell, mucinous, and transitional tumors8-10. Apart from clear-cell carcinomas, type I tumors are indolent and restricted to the ovary in early stages9,10. On the other hand, the type II, which make up nearly 75% of all EOCs, are more aggressive, progress very rapidly, and are detected at advanced stages. Type II tumors originate from the fallopian tube and include high-grade serous carcinoma, high-grade endometrioid carcinosarcoma, and undifferentiated carcinoma9,10. The key difference between type I and II tumors is the high chromosomal instability in the latter. Molecular-based studies have also revealed that type II tumors have a relatively higher frequency of mutations inactivating tumor protein 53 (TP53), which rarely occurs in type I tumors (Figure 1). This implies that TP53 is more genetically stable in type I compared to type II tumor10. TP53 is a tumor-suppressor gene that codes for a checkpoint protein responsible for regulating the cell cycle and evaluating DNA damage. If damaged DNA is repairable, it signals DNA-repair genes such as BRCA1, but if the damage cannot be repaired, TP53 induces an apoptotic signal in the cell, killing it, and thereby preventing tumor formation4,10. In cancers such as breast, bone, leukemia, and sarcomas, frequent TP53 mutations were observed. The majority of the TP53 mutations are missense mutations resulting in single amino acid substitutions10. The mutation patterns from an abnormal TP53 can be used to infer the possible mutagens and molecular pathogenesis responsible, thus making this gene a unique avenue of exploration for cancer studies10. Abnormalities in homologous recombination repair, retinoblastoma protein, cyclin E1 and NOTCH3 pathways are also observed in type II carcinomas. In addition to genetic alterations in BRCA1-mutant type II tumors, the expression of BRCA1 can also be perturbed by promoter methylation (Figure 1)10.
Table 1. Ovarian cancer cell types and their characteristics. The table was adapted from (2).
|
Ovarian cancer cell type |
Characteristics |
|
Surface epithelial cells |
|
|
Serous |
Cystic neoplasms that can be either low-grade or high-grade carcinoma. Commonly found bilaterally. |
|
Mucinous |
Larger cystic masses than serous tumors. Contain gelatinous fluid, rich in glycoprotein. |
|
Endometrioid |
Comprises solid and cystic material containing glands that resemble endometrial epithelium. |
|
Clear cell |
Rare type of ovarian cancer. Comprises epithelial cells with abundant clear cytoplasm. |
|
Germ cells |
|
|
Mature teratoma |
Benign teratoma (dermoid cyst) that is lined with skin. Tumor may contain hair, teeth, bone, thyroid, and neural tissue. |
|
Yolk sac tumor |
Known as endodermal sinus tumor. Derived from germ cells from the extraembryonic yolk sac lineage. |
|
Non-gestational choriocarcinoma |
Resembles placental tissue. May occur in prepubertal females as a result of an ectopic pregnancy after this age. |
|
Dysgerminoma |
The counterpart of testicular seminoma. Tumor consists of cells with clear cytoplasm and central nuclei. |
|
Sex cord-stromal cells |
|
|
Granulosa |
Tumor of granulosa cells that secrete large amounts of estrogen. |
|
Fibroma |
Composed of fibroblasts and is hormonally inactive. |
|
Thecoma |
Tumor of theca cells. Composed of lipid droplets and is hormonally active. |
|
Fibrothecoma |
Tumor composed of both fibroma and thecoma components. |
|
Sertoli-Leydig |
Tumors that resemble granulosa cells but cause defeminization due to secretion of testosterone. |
Table 2. List of probable genes associated with the pathogenesis of ovarian cancer. Retrieved from: https://ghr.nlm.nih.gov/gene, http://www.genecards.org
|
Gene symbol |
Name |
Function |
Signaling pathway |
|
KRAS |
KRAS, proto-oncogene, GTPase |
Involved primarily in regulating cell division. |
RAS/MAPK |
|
BRAF |
Threonine-protein kinase B-Raf |
BRAF helps transmit chemical signals from outside the cell to the cell's nucleus. |
RAS/MAPK |
|
ERBB2 |
ERB-B2 receptor tyrosine kinase 2
|
Epidermal growth factor receptor family of receptor tyrosine kinases. |
MEMO1-RHOA-DIAPH1 |
|
CTNNB1 |
Catenin, beta 1
|
Responsible for cell adhesion and communication |
WNT |
|
PTEN |
Phosphatase and tensin homolog
|
Enzyme that regulates cell migration, adhesion, angiogenesis and triggers apoptosis. |
PI3K |
|
PIK3CA |
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
|
Phosphorylates signaling molecules, triggering a cascade of reactions contributing to cell division, migration, and growth. |
PI3K |
|
ARID1A |
AT-rich interaction domain 1A
|
Encodes for a subunit of SWI/SNF protein complexes that assist in regulating cell proliferation. |
AMPK |
|
PPPR1A |
Protein phosphatase 1, regulatory (inhibitor) subunit 1A |
Elevates intracellular cAMP, increases I-1 activity in many tissues. |
cGMP-PKG |
AMPK: AMP-activated protein kinase; cGMP-PKG: cGMP-dependent protein kinase. MAPK: mitogen-activated protein kinase; PI3K: phosphoinositide 3-kinase; RAS: subset of the RAS small GTPase superfamily; MEMO1: mediator of cell motility 1; RHOA: ras homolog family member A; DIAPH1: diaphanous related formin 1.
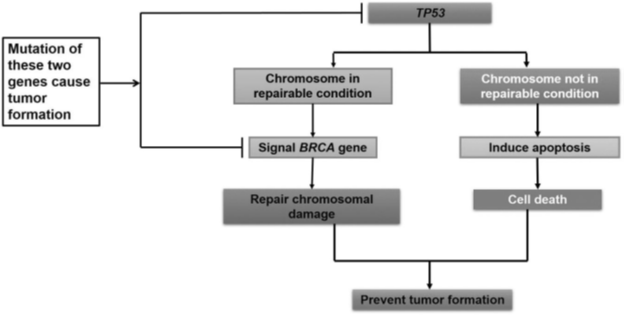
Figure 1: Schematic diagram for the action of Tumor Protein 53 (TP53) gene and Breast Cancer (BRCA) gene. TP53 can assess if the damage on a chromosome is repairable. If it is, it can signal the BRCA gene, which activities a chromosome-repairing mechanism to restore the proper functioning of the target chromosome. In an event where the chromosome is not repairable, TP53 induces apoptosis of the cell. Both these pathways prevent tumorigenesis. Because this mechanism requires two gene products (TP53 and BRCA), mutations in either of these gene results in a dysfunctional pathway, promoting tumor formation.
Epigenetic Modifications
Epigenetic changes contribute to disease progression and malignant transformation that may result in ovarian cancer cells being resistant to certain drugs. Epigenetic modification is the temporary change of gene expression as a result of several external factors12. Epigenetic alterations are one of the main reasons for the emergence of drug-resistant cancer cells, which is a major issue affecting disease remission. Epigenetic modifications are potentially reversible, which opens the possibility of therapeutic interventions. These modifications can affect chromatin structure and gene regulation, which are important characteristics of cancer formation. Cellular functions such as cell differentiation, cell-cycle progression, apoptosis, and gene expression can be severely affected by several different epigenetic changes, such as but not limited to DNA methylation, histone modifications, and chromatin remodeling and repressive complexes. Some of the notable epigenetic modifications that are linked to ovarian cancer are discussed below.
NOTCH Signaling Pathway
This pathway is responsible for governing binary cell fate, patterning, proliferation, growth, and programmed cell death13. Dysfunctional NOTCH signaling is known to promote oncogenic effects by inhibiting apoptosis while accelerating cell proliferation. Abnormal NOTCH signaling alters the developmental state of a cell, allowing the cell to adopt a continuous proliferative nature making it cancerous14. The NOTCH developmental pathway in mammals includes the NOTCH receptors (NOTCH1-4), delta ligands (DLL1, DLL3, and DLL4), and serrate ligands (JAGGED1 and JAGGED2)13. Epigenetic changes, such as hypermethylation and hypomethylation of NOTCH genes, have been linked to ovarian cancer. Lack of DLL1 expression caused by promoter hypermethylation is known to down regulate NOTCH1 expression, ultimately leading to gastric cancer14. Numerous studies have identified NOTCH3 to be a candidate oncogene. In comparison to normal ovarian tissue, NOTCH3 expression was preeminent in ovarian cancer tissue15,16. Cell-cycle regulator cyclin-dependent kinase inhibitor 1 (CDKN1), related to cell proliferation, is a target gene of NOTCH1 and is known to interact with the p53 pathway to manipulate the cell cycle13,15. The NOTCH pathway is also involved in reducing histone H3K4 methylation and down-regulating the retinoblastoma tumor-suppressor gene (RBF) gene. Such epigenetic changes have also been linked to other human cancers13. Using real-time polymerase chain reaction (RT-PCR), Jung et al. compared the expression of NOTCH3, JAGGED1 and JAGGED2 mRNAs between ovarian carcinomas and benign tumors. Their study detected a 3-fold and 10-fold increase in expression of JAGGED1 and JAGGED2 respectively in serous carcinomas17. Further studies regarding the involvement of the NOTCH pathway in ovarian oncogenesis are in progress, and its implication in ovarian cancer has yet to be fully understood17.
Forkhead Box M1 (FOXM1)
FOXM1 plays an important role in cell-cycle progression, apoptosis, angiogenesis, and DNA-damage repair18-2-. During the cell cycle, FOXM1 regulates the G1/S and G2/M phases and maintains the integrity of the spindle fibers during mitosis. In numerous cancer types, such as ovarian, breast, prostate, lung, and gastric, it has been observed that the 12p13 chromosomal band containing the FOXM1 gene is amplified21. Epigenetic modification of FOXM1 is carried out by lymphoid specific helicase (HELLS). The modification is known to change the pattern of methylation of FOXM1 gene resulting in inducing malignant transformation by disrupting epithelial differentiation in normal cells22. Using microarrays, a systemic analysis of gene expression demonstrated the up regulation of FOXM1 mRNA in several ovarian tumors23. The up regulation of FOXM1 leads to proliferation, migration, and invasion in breast, pancreatic, and gastric cancer. Up-regulation in extracellular matrix-degrading factors and angiogenic factors, such as urokinase-type plasminogen activator (uPA), uPA receptor, matrix metallopeptidase 2 and vascular endothelial growth factor are also observed in these cancer types21. It has been hypothesized that FOXM1 is a crucial regeneration factor for epithelial tissues after injury21. When overexpressed, FOXM1 is known to up-regulate pluripotent gene expressions such as octamer binding transcription factor 4, homeobox protein NANOG, and sex determining region Y-box 224. Some of the FOXM1-induced differentially methylated genes include those that are hypermethylated such as: Chromosome 6 open reading frame 136 (C6orf136), mannosyl (alpha-1,3-)-glycoprotein beta-1,2-N-Acetylglucosaminyltransferase (MGAT1), NADH:ubiquinone oxidoreductase subunit A10 (NDUFA10), and platelet-activating factor acetylhydrolase 1b catalytic subunit 3 (PAFAH1B3). On the other hand, genes such as signal peptidase complex subunit 1 (SPCS1), filamin A alpha (FLNA), chondroitin polymerizing factor (CHPF), and glycosyltransferase 8 domain containing 1 (GLT8D1) were found to be hypomethylated as a result of FOXM122. FOXM1 is further capable of initiating an oncogenic pathway by predisposing stem progenitor cells to tumorigenesis25. According to a study from the Genome Atlas Consortium, FOXM1 elevation may lead to the growth of pathological EOC26. FOXM1 hyperactivity is a steady feature of metastasis and proliferation of EOCs27,28.
WNT Signaling Pathway
The WNT signaling transduction pathway is essential for embryonic development and tumorigenesis29. It is responsible for proper follicular development and ovarian function. Overactivation of this signaling pathway has reportedly led to colon, liver, cervical, and ovarian cancers30. Zhu et al. observed the effects of Dickkopf WNT signaling pathway inhibitor 2 (DKK2) on WNT/β-catenin signaling in ovarian cancer cells by evaluating the β-catenin staining in SKOV3 and ES-2 cells31. In their study, down-regulation of β-catenin expression in DKK2-transfected cells was noted when compared with untreated cells. In addition, luciferase reporter assay in SKOV3 and ES-2 cells showed that T-cell factor (TCF) activity was significantly blocked in cells transfected with DKK231. Therefore, it appears that DKK2 participates in the negative feedback loop that inhibits WNT signaling. Zhu et al. also found that cells transfected with DKK2 had poor expression of MYC proto-oncogene, BHLH transcription factor (c-MYC) and CDKN1. To further investigate this, the expression of focal adhesion kinase (FAK), MMP2 and MMP9 were measured using western blotting. The results showed FAK and MMP2 expression were lowered in DKK2-transfected cells31.
WNT signals are known to be transduced in two main ways: β-Catenin-dependent and independent. During β-catenin-dependent transduction, frizzled family receptor proteins provides an anchor for the WNT ligand to stabilize the β-catenin for proper translocation into the nucleus30. Cytoplasmic protein, β-catenin, is a component of cell–cell adhesion junctions and a mediator of the WNT signaling pathway. In normal cells, phosphorylation facilitated β-catenin degrading occurs through the ubiquitin pathway32. Nuclear β-catenin is overexpressed in ovarian cancer as a result of up-regulation of the β-catenin gene itself, and due to the phosphorylation of glycogen synthase kinase 3β (GSK3β) which is a component of the β-catenin destruction complex33. Mutation of this ubiquitin pathway or the β-catenin destructive complex causes nuclear accumulation of β-catenin which stimulates the transcription and translation of approximately 23 genes involved in ovarian cancer proliferation and growth30,32. E-cadherin is activated by the binding of β-catenin to TCF and upregulated in invasive ovarian cancer cells32. In addition, TCF is also known to activate genes such as c-MYC and CDKN1, assisting in the initiation and progression of ovarian tumor30,33.
Epithelial–mesenchymal Transition (EMT)
The EMT is recognized as a prerequisite for metastasis since it allows cancer cells to become invasive. This transition can be stimulated by several factors such as transforming growth factor beta (TGFβ), bone morphogenic proteins (BMPs), receptor tyrosine kinases, WNT and NOTCH signaling pathways. These signals influence EMT-inducing transcription factors such as Snail (SNAI1), Slug (SNAI2), Twist-related protein 1 (TWIST), and zinc finger transcription factor (ZEB1/2)34. During the process of EMT, ovarian cancer cells transition from an epithelial to a mesenchymal morphology while apical and basolateral epithelial cell-specific tight junction proteins, such as E-cadherin and cytokeratin, are down-regulated. On the contrary, expression of vimentin gene and mesenchymal molecule N-cadherin are observed35. The EMT mechanism relies on the intrinsic ability of most embryonic epithelial cells to exhibit plasticity and alter their cell–cell contact structures, leading to branching morphogenesis or delamination36. Down-regulation of E-cadherin during EMT is a key factor for the invasive and metastatic nature of ovarian clear cell carcinoma. According to Takai et al., expression of E-cadherin reduces as the ovarian cancer increases in stage. The reduction of E-cadherin during stage I was 30.4% and rose to 52.7% reduction in stage IV35. EMT is exploited to increase cancer cell aggressiveness through acquiring chemoresistance, regenerative properties, and developing the ability to escape host immune cells36. Fang et al. found that in order to sustain the EMT characteristics of ovarian cancer cells, the presence of SOS RAS/RAC guanine nucleotide exchange factor 1 (SOS1)/epidermal growth factor receptor kinase substrate 8 (EPS8)/Abl interactor 1 (ABI1) complex is necessary. This complex supports the invasiveness of ovarian cancer cells and loss of any component of this complex reduces mesenchymal-like traits and restores epithelial characteristics to ovarian cancer cells34.
DNA Methylation
Most of the DNA methylation takes place at CpG dinucleotides, which can be sporadically distributed throughout the genome or located in CpG-rich regions called CpG islands37,38. Normal cells exhibit global hypermethylation of repetitive and satellite sequences accompanied by locus-specific hypomethylation at active gene promoters and enhancers, which regulates the expression of antitumor genes37,38. In cancer cells, global hypomethylation accompanied by gene-specific hypermethylation can cause aberrant expression of tumor-suppressor genes or oncogenes. DNA-methylation patterns are widely known to affect gene expression in ovarian cancer. Gene promoter regions, gene bodies, and locations several kilobases upstream or downstream of genes all contain CpG islands38. When these CpG islands are methylated in error, it can cause silencing of tumor-suppressor genes. Some of the genes silenced by methylation in ovarian cancer include PTEN39, BRCA40, paternally expressed gene 3 (PEG3)41, RAS-associated domain family member 1 (RASSF1)42, and mutL homolog 1 (MLH1)43,44. Further details about these genes are discussed below.
PTEN. PTEN is an antitumor gene, and the mutation of this specific gene is observed in ovarian cancer45. PTEN participates in the negative regulation of PTEN/phosphatidylinositol 3-kinase (P13K)/protein kinase B (AKT) pathway that regulates the cell cycle, cell division and apoptosis. PTEN is also proposed to induce drug resistance via the P13/AKT pathway in ovarian cancer cells45,46. Dysfunction of PTEN protein activated P13K/AKT pathway resulting in ovarian carcinoma becoming resistant to cisplatin45. PTEN also contributes to the drug resistance of ovarian cancer cells as a result of DNA methylation46. Studies suggest that methylated genes are involved in the process of ovarian carcinoma drug resistance, and PTEN could be a noteworthy regulator45,46.
BRCA. Multiple studies reveal that many EOCs harbor somatic mutations in BRCA1 or BRCA247,48, however, it was found that BRCA proteins are also subject to epigenetic modifications48. DNA-damage repair, transcription, cell-cycle checkpoint regulation, protein ubiquitination and apoptosis are some of the critical functions of BRCA genes49. Due to mutated or non-functional BRCA1 and BRCA2, double-stranded DNA breaks are not repaired, thus increasing the possibility of more mutations50. Studies performed to elucidate BRCA mutations revealed that these mutations cause the loss of checkpoint proteins in the cell cycle, leading to chromosomal instability51,52. BRCA deficiency is being studied using DNA microarray-based gene-expression analysis to understand its ability to manipulate gene expression53. The changes in protein expression as a result of BRCA mutations were thoroughly investigated by studying patients with ovarian carcinoma. The study by Gau et al. explored the different ways by which a deficiency of BRCA1 modulated ovarian cancer progression by regulating the expression of cell motility proteins54. They found differential expression of proteins involved in actin cytoskeletal/cell adhesion remodeling in BRCA1 deficiency, which also correlated with higher stages of ovarian cancer54. Functional BRCA1 is involved in maintaining optimal expression of regulatory proteins for the cell cytoskeleton, cell adhesion and migration in EOC. The amount of expression varies between early- and advance-stage tumors48. Chan et al. found a distinguishing feature between BRCA1 and BRCA2 inactivation whereby epigenetic silencing by promoter hypermethylation results in a significant loss of function only for BRCA148.
RASSF1. Hypermethylation of the tumor-suppressor gene RASSF1A leads to gene inactivation, which is an early event in ovarian cancer development55-57. Methylation of CpG islands in the promoter of RASSF1A gene results in its epigenetic inactivation58. When compared to a control group with only 0-21% promoter methylation of RASSF1A, the frequency of this methylation in patients with cancer was revealed to be 30-58%56. RASSF1A regulates multiple functions such as apoptosis, tubulin dynamics and cell-cycle arrest in cancer cells59. RASSF1A promoter methylation is a common epigenetic inactivation which is implicated in human cancer and it results in silencing of RASSF1A expression57-59. Hypermethylation of RASSF1A promoter is correlated with ovarian cancer and can be important biomarker in detecting the carcinogenic process of ovarian cancer56.
PEG3. PEG3 is one of several imprinted genes which play a vital role in human oncogenesis60. These imprinted genes are very susceptible to loss of function compared to tumor-suppressor genes because such genes can lose their function as a result of inactivation of just one functional allele61. PEG3 is crucial in the p53/c-MYC mediated apoptotic pathway, and, therefore, down-regulation of imprinted genes becomes a key factor in ovarian oncogenesis62. It is anticipated that re-expression of PEG3 would inhibit survival and proliferation in human ovarian carcinogenesis60. There is prediction that PEG3 is highly associated with carcinogenesis if the promoter of PEG3 was methylated accordingly63. Therefore, it was concluded that the loss of PEG3 function as a result of promoter methylation may lead to pathogenesis of multiple types of ovarian cancers63.
MLH1. Promoter hypermethylation of MLH1, an epigenetic inactivation of the MMR system resulting in loss of MMR, was recognized as a potential event for ovarian cancer pathogenesis64,65. Studies revealed a staggering frequency of MLH1 promoter hypermethylation, ranging from 6% to 12.5% in ovarian cancer66. Previous studies also revealed MLH1 deficiency was linked to resistance to cisplatin and carboplatin67,68. The mRNA expression of MLH1 appeared to be inversely correlated with its DNA methylation level, revealing the importance of cytosine methylation in controlling MLH1 expression69. These findings confirmed that down-regulation of MLH1 is possible by Histone H3K27 methylation and histone deacetylation. Down-regulation of MLH1 is also a possible way ovarian cancer progresses by further drug resistance67,68. The methylation rate in certain genes are linked to the degree of clinical pathogenesis of ovarian cancer. It has been demonstrated that methylation in secreted frizzled-related protein (SFRP)1, -2, -4 and -5; SOX1; paired box 1 (PAX)1 and LIM homeobox transcription factor 1 α (LMX1A) were the highest in those with more aggressive forms of ovarian carcinoma and declined progressively in borderline malignancy70,71.
Hypomethylation
The process of hypomethylation in cancer has been less studied than hypermethylation. Hypomethylation at repeat elements is a major cause of genomic instability and epigenetic de-repression of oncogenes in ovarian cancer. There are several examples of genes overexpressed by hypomethylation. Some of these genes include methylation-controlled-DNAJ Gene (MCJ)71,72, synuclein-γ73,74, taxol-resistant-associated gene 3 (TRAG3)75, and brother of the regulator of imprinted sites (BORIS)76. A class of repetitive sequences called retrotransposons, specifically long interspersed elements, short interspersed elements, and long terminal repeat retrotransposons are hypothesized to be targets for hypomethylation70,77. EOC histotypes exhibit specific differences in global hypermethylation lacking any predictive hypomethylation patterns78. Hypomethylation is generally correlated with increasing stage of cancer, which is consistent with the paradigm that DNA hypomethylation is a key oncogenic factor. It is suggested that the degree of hypomethylation of repeat sequences increases from benign precursors to malignant tumors78.
Gene Hypomethylation and Ovarian Cancer Pathogenesis
The pathogenesis of ovarian cancer involves hundreds of genes. Past research has established that the hypomethylation of several genes is a major cause in cancer initiation.
Solute carrier family 6 member 12 (SLC6A12). Sung et al. identified up-regulation of the neurotransmitter transporter, SLC6A12, as a contributor to ovarian cancer metastasis79. This gene product functions in the uptake of gamma-aminobutyric acid (GABA) and betaine in the liver, kidneys, and brain surface80. SLC6A12-transfected human ovarian cancer (SKOV3) cells showed a two-fold upsurge in cell migration when compared to cells transfected with enhanced green fluorescent (EGF) protein cDNA79. Additionally, overexpression of SLC6A12 increased the invasive activity of SKOV3 cells. Regulation of SLC6A12 was found to be influenced by hypomethylation of the promoter region at 18 CpG sites. In metastatic tumor cells, DNA methylation of these sites was significantly reduced in comparison to injected ovarian carcinoma cells. However, gene expression was shown to be enhanced when treated with a DNA methyltransferase inhibitor or histone deacetylase inhibitor79,80.
Carbonic anhydrase (CA9). Like SLC6A12, CA9 hypomethylation also affects the pathogenicity of ovarian cancer. The expression of membrane-associated glycoprotein CA9 serves as an indication of tumor hypoxia. Sung et al. observed that hypomethylation resulted in overexpression of CA9, increasing the invasive and migratory capacity of metastatic SKOV3 cancer cells81.
Transmembrane protein 88 (TMEM88). The genes TMEM88 and Msh homeobox 1 (MSX1) have been found to result in platinum resistance and thus increased pathogenicity via promoter hypomethylation82. TMEM88 regulates the canonical WNT signaling pathway, which causes the accumulation of β-catenin in the cytoplasm and its translocation into the nucleus82. De Leon et al. observed an increase of TMEM88 expression in platinum-resistant tumors. Hypomethylation of the promoter CpG islands resulted in an increase in gene expression. The researchers suggested that platinum-based therapy likely causes promoter hypomethylation, leading to up-regulation of TMEM8882. Increased expression of TMEM88 inhibits WNT signaling, halts cell proliferation, and allows the cancer cells to enter a state of dormancy. During this state, cancer cells attain resistance to platinum therapies, eventually producing a lineage of resistant tumor cells82.
MSX1. During the process of embryogenesis, MSX1 is responsible for epithelia–mesenchymal interactions83. Researchers observed that in cancer, down-regulation of MSX1 was linked to decrease in six out of eight CpG methylations83,84. Cell lines that exhibited platinum resistance had lower MSX1 gene expression in comparison to those that were platinum sensitive. Therefore, the researchers concluded that hypomethylation of the intragenic CpG sites was associated with resistance to platinum chemotherapy84. MSX1 expression is of specific importance for the treatment of High-Grade Serous Epithelial Ovarian Cancer (HGSOC). Although, ovarian cancer is responsive to platinum-based chemotherapy, it has been noticed that HGSOCs are particularly resistant to such treatment84. Studies revealed and identified CpG sites that were hypomethylated in MSX1 gene resulting in its down regulation in HGSOCs which resulted in its unique insensitivity to platinum-based drugs. Understanding the epigenetic modifications in genes such as MSX1 unlocks different avenues to explore other suppressor genes to elucidate their relation to possible chemoresistance of different types of ovarian cancers to chemotherapeutic agents83,84.
Preferentially expressed antigen in melanoma (PRAME) and cancer/testis antigen 45 (CT45). Ovarian cancer pathogenesis can also be affected by the PRAME gene. PRAME is a cancer-testis antigen that is specific for tumors. Due to these characteristics, cancer-testis antigens have opened the possibility of developing antigen-specific cancer vaccines. Oehler et al. observed DNA hypomethylation of the PRAME promoter in both EOC and in the high-grade serous subtype of ovarian cancer85. Hypomethylation was correlated with increased PRAME expression, as EOC with high levels of PRAME expression were seen to have a reduction in methylation. Additionally, promoter and the transposable element, LINE1 hypomethylation (a global methylation status surrogate) were associated, suggesting that hypomethylation commonly observed in EOC is connected to PRAME regulation85,86. Similarly, the CT45 gene is also related to cancer pathogenesis. This gene is an X-linked CT gene associated with oncogenesis. In endothelial cancer cells, CT45 promoter activity was directly repressed by DNA methylation, compared to the control cells where CT45 was hypermethylated86.
Histone Modification
Chromatin is an important component of the physiological template of eukaryotic genetic information and is composed of DNA, histones, and other proteins condensed into protein complexes. It is an organized structure that consist of nucleosome repeats joined by linker DNA, called H187. Nucleosomes are composed of histone octamers each consisting of two core histones: H2A, H2B, H3 and H4 wrapped by 147 base pairs of DNAs78,88. The post-translational modifications of the histone tails such as methylation, acetylation, ubiquitination and sumoylation are catalyzed by specific enzymes resulting in changes in gene expression. Histone modifications also affect accessibility to transcription factors, repressors and proteins that regulate DNA repair and replication88. Methylation of lysine residues can have different consequences depending on which histone subunit is affected and on its location within the histone tail. Methylation of histone 3 lysine 27 (H3K27me), histone 3 lysine 9 (H3K9me), or histone 4 lysine 20 (H4K20me) are linked to repression of gene expression by causing the compact state of chromatin87,88. Di- and tri-methylation of histone 3 lysine 4 (H3K4me2 or me3) are strictly regulated by histone acetyltransferases (HATs) and deacetyltransferases (HDACs)87-89. Acetylation of lysine residues in histone tails lead to a flexible chromatin structure that is more available to transcription factors increasing gene expression; whereas, deacetylation makes it difficult for transcription factors to access genes, causing transcriptional repression89.
In ovarian cancer, abnormal HDAC pathways have been implicated in promoting cancer growth and metastasis88. HDAC enzymes are classified into four classes: I, II, III and IV, each with multiple members that have diverse expression profiles. Class III HDAC family members are sirtuin proteins (SIRT1-7) which are nicotinamide adenine dinucleotide (NAD+)-dependent enzymes. The other three classes are zinc dependent. Mammalian homologs of the yeast silent information regulator (ir2), sirtuins can act as HDAC and as a deacetylase for p53 which belongs to non-histone proteins88. Jang et al. observed that SIRT1 expression was elevated in malignant EOC compared to benign tumors. Their study also revealed that SIRT1 up-regulation in a specific subgroup of malignant serous EOC was correlated to its increased survival capability90.
Caslini et al. studied how histone modifications affected expression of GATA transcription factors on five ovarian cancer cell lines known as human immortalized ovarian (HIO) surface epithelial HIO-117, HIO-114, A2780, SKOV3, and ES2 cells91. GATA transcriptional factors are crucial in determining lineage during cell differentiation. Loss of GATA factors (GATA4, GATA5, GATA6) are reported in various cancer types, including ovarian cancer. GATA4 expression is reduced and GATA6 is lost from the nucleus in 85% of ovarian tumors. The study concluded that GATA transcription factors can be silenced by altered histone modification of the promoter loci inhibiting the expression of tumor suppressor disabled-2 (DAB2)91. Genome-wide studies revealed that genetic alterations affecting expression of histone-modifying genes are present in various human cancer types including, ovarian carcinomas58. Table 3 lists histone-modifying genes that can be altered in ovarian carcinoma.
Table 3. Various histone modification genes altered in ovarian cancer (OC). This table was adapted and slightly modified from (87).
|
Type |
Common name |
Alteration in OC |
|
Histone deacetylases: |
Histone deacetylase 1 (HDAC1) |
Up-/down-regulation |
|
Histone acetyl transferases: |
E1A binding protein p300 (P300) |
Mutation, translocation, deletion |
|
CREB binding protein (CRB) |
Mutation, translocation, deletions |
|
|
General control non-depressible member 5 (GCN5) |
Down-regulation, mutation |
|
|
Histone acetyltransferase binding to HBO1 |
Up-regulation |
|
|
Histone methyltransferase: |
Suppressor of variation 3-9 homolog (SUV39H1-2) |
Mutation overexpression |
|
Enhancer of zest homolog 2 (EZH2) |
Amplification, up-regulation |
|
|
SUZ12 Polycomb repressive complex 2 subunit (SUZ12) |
Up-regulation |
|
|
Polycomb ring finger (BMI1) |
Up-regulation |
EZH2 and Polycomb Repressive Complexes PRC2 and PRC1
EZH2 can epigenetically silence the expression of its target genes by catalyzing lysine 27 methylation on histone H392. The catalytically inactive EZH2 is activated by the components of PRC293. The target genes of PRC2/EZH2 are involved in pathways that control stem cell differentiation, proliferation, and tumorigenesis94. EZH2 expression is elevated in high-grade serous EOC, and in tandem with PRC2 components EZH2 regulates cancer cell proliferation, invasion and apoptosis95. When compared to non-cancerous cells, some components of PRC2, such as SUZ12, are overexpressed in EOC93,95,96. The up regulation of EZH2 in endothelial ovarian cancer is linked to stimulation of paracrine vascular endothelial growth factor. Further investigation found that EZH2 knockdown inhibited angiogenesis and reduced cell proliferation96. The pro-apoptotic gene Harakiri (HRK) is silenced by H3K27me3 methylation in EOC cells95. EZH2 inhibitors are essential in reducing H3K27me3 in cancer cell lines. Cell lines with wild-type EZH2 are usually less sensitive to EZH2 methyltransferase inhibitor GSK12695,96. These findings led to the proposal that methyltransferase-independent function of EZH2 is also associated with malignant phenotypes seen in cancer cells with up-regulation of EZH2. Knockdown of EZH2 can inhibit PRC2, which can ultimately reduce the growth of EOC cells dependent on elevation of PRC292,94. Therefore, EZH2 inhibitors can be evaluated based on their efficacy in reducing EOC growth in preclinical models93,95,96. Furthermore, manipulating the methyltransferase activity of EZH2 and targeting PRC2 complex formation can provide alternative tactics for targeting cancer cells96.
SWI/SNF Chromatin Remodeling Complexes and Ovarian Cancer
The SWI/SNF chromatin remodeling complexes are involved in cancer initiation and progression97. Mutations in at least one member of the SWI/SNF complex is common in human tumors98. SWI/SNF complexes control crucial cellular processes, such as cell cycle, apoptosis, cell differentiation, genomic stability, and DNA repair97. Based on the presence of ARID1A/B or ARID2 and polybromo 1 (PBRM1) subunits, the SWI/SNF complex can be further classified into BRG1-associated factor (BAF) or polybromo-associated BAF complex (PBAF) respectively99. In ovarian cancer, mutations in several SWI/SNF complexes were found100,101. These mutations lead to altered nucleosome positioning, making it difficult for RNA poly 11 or other transcription factors to access DNA102. Through extensive genomic studies, three important limitations in tumor suppression mechanisms by BAF complex were found102,103. Firstly, the fundamental mechanism gives rise to genetic supremacy103. Secondly, in-vitro chromatin remodeling assays failed to recreate this mechanism103. Thirdly, this mechanism is only present in a highly specific tumor environment103. The involvement of SWI/SNF complex in regulating chromatin organization, RNA transcription, DNA damage repair and cell division make it extremely difficult to elucidate its role in cancer development100,102. Two important subunits of the SWI/SNF complex are further discussed below.
ARID1A. The ARID1A is a component of the ARID1 subunit of the SWI/SNF complex which can mobilize nucleosomes and subsequently regulate epigenetics of many genes including those that are responsible for ovarian clear-cell carcinoma, an aggressive form of ovarian cancer known to be resistant to typical chemotherapy. Chromosomal translocation of tumor-suppressor ARID1A gene has been linked to breast and lung cancer, whereas leukemia cells with silenced ARID1A resulted in them being resistant to Fas-mediated apoptosis104. Abnormal chromosome remodeling complex and the loss of BAF250a expression of protein are some of the known effects of ARID1A inactivation105. Transcriptome sequencing has allowed identification of frequent mutation of the ARID1A gene in endometroid and clear-cell ovarian carcinomas. Abnormality in the ARID1A gene is a consistent feature in cancer, such as gene rearrangement in breast cancer and gene deletion in lung cancer. This suggests that ARID1A has tumor-suppressor properties105. In one study, 46% of ovarian clear-cell carcinomas and 30% of endometrial carcinomas were found to have ARID1A mutation using exon resequencing105,106. ARID1A mutations were mostly observed in atypical endometriosis and primary malignant lesions107. Mutation of chromatin-modifying genes is a type of epigenetic change which can result in tumorigenesis. Comparative analyses of exon sequences and immunoaffinity purification indicate the link of four gene mutations to ovarian clear-cell carcinoma. These genes include PIK3CA, KRAS Proto-Oncogene (KRAS), Protein Phosphatase 2 Scaffold Subunit Alpha (PPP2R1A), and ARID1A104,105. Lowery et al. studied 212 ovarian cancer samples and observed BAF250a loss in 45% of the clear-cell samples and in 41% of the endometroid samples105. This suggests that the loss of BAF250a protein is a pathological factor in the genesis of ovarian clear-cell and endometroid cancer105,107. Mutation in the ARID1A gene may encode for a protein like BAF250a but nonfunctional. However, mutated ARID1A inactivates the chromatin remodeling pathway, leading to cancer105,107.
SWI/SNF-related, Matrix-associated, Actin-dependent Regulator of Chromatin, Subfamily A, Member 4 (SMARCA4). SMARCA4 is a chromatin remodeling gene which when mutated becomes inactivated and contributes to the formation of small cell carcinoma of the ovary, hypercalcemic type (SCCOHT)108. SCCOHT is primarily seen in young women, but it is a rare and extremely aggressive type of ovarian cancer. The characteristic features of SCCOHT include abnormal layers of small cells with limited cytoplasm, hyperchromatic nuclei, and small nucleoli. It is suggested immature ovarian cells may be the precursor for SCCOHT108. Patients with SCCOHT have a long-term survival rate of only 33% and typically do not live over 2 years after diagnosis, regardless of the cancer stage109. Ramos et al. investigated the pathogenesis of SCCOHT and found that 69% of the samples contained SWI/SNF mutations and 82% had a loss of SMARCA4110. Jelinic et al. found that Inactivation through bi-allelic SMARCA4 mutation is a major determinant of SCCOHT108. SMARCA4 inactivation was caused by frameshift, nonsense, and splice-site mutations108,109. A mutation that causes inactivation in one of the subunit genes results in the loss of tumor-suppression ability of the SWI/SNF complex, leading to uncontrolled cell proliferation and resulting in SCCOHT109. In a study to assess the loss of SMARCA4 expression in ovarian tumors, it was found that 42 samples out of 46 lacked SMARCA4 expression which is approximately 91%. In addition, the four samples that did not exhibit the loss of SMARCA4 expression lacked a different protein which is also a component in forming the SWI/SNF complex108-110.
Role of Micro RNAs (miRNAs)
miRNAs are non-protein coding RNA sequences between 15 to 22 nucleotides in length111. Since miRNAs are such short sequences, they do not have to be a perfect complement in order to bind their target mRNA. This means multiple miRNAs can bind to one target mRNA and in turn, one miRNA is able to control multiple mRNA targets112. Specific miRNAs have been shown to be either down regulated or up regulated depending on the type of cancer. Some of these miRNAs when up-regulated, can function as oncogenes by inactivating tumor-suppressor genes. In contrast, when down-regulated, they can serve as tumor-suppressor genes by negatively affecting oncogenes112,113. Numerous studies have been performed to see if specific miRNAs dysregulation patterns can be detected in ovarian cancer. The Cancer Genome Atlas project analyzed 500 high-grade serous ovarian carcinoma and documented the variations in their miRNA levels113. Other studies narrowed the pool further by comparing miRNAs in normal ovaries to miRNAs in ovarian cancer. Out of 310 dysregulated miRNAs discovered across multiple studies, about 34 were seen to be consistently dysregulated in ovarian cancer cells. miR-127 and miR-31 were consistently down-regulated, whereas miR-20a and miR-200b/c was up-regulated113.
The expression level of a specific miRNA can provide information regarding the aggressiveness of the cancer111. Certain miRNAs may indicate the type of tissue from which the cancer originated, allowing treatment options to be narrowed down. The dysregulation of a specific miRNA, such as an increase in miRNA-17 production, has been noted in patients with chemo-resistant colorectal cancer112. Elevated miRNA-17 has also been correlated with an increase in the invasion and proliferation of breast cancer112. To explore this relationship between miRNAs and cancer, Lui et al. studied the interaction between liver kinase protein (LKB1), which functions by regulating p53 and p21/WAF1111. They found that in presence of miR-17, transcription and translation of LKB1, p53, and p21 in ovarian clear-cell carcinoma were reduced. miR-17 is known to affect cell-cycle regulation and stimulate the proliferation and invasion of ovarian clear-cell carcinoma111. They also found that the absence of functional miR-17 caused tumor suppression. Therefore, they concluded that miR-17 interferes with LKB1–p53–p21/WAF1, leading to oncogenesis111.
It was found that miR-215 was down-regulated in EOC cell lines and associated with lymph node metastasis112. NIN/RPN12 binding protein (NOB1) was identified as a target gene of miR-215. Critical oncogenic pathways through activation of the Mitogen-activated protein kinases (MAPK) pathway and p38MAPK phosphorylation were observed as a result of miR-215 up-regulation111,113. Table 4 shows a representative list of different miRNAs involved in dysregulation in EOC. Expression patterns of miRNAs can be used as biomarkers for novel ovarian cancer treatment methods114. It was seen that patients with up-regulation of miR-182 displayed reduced sensitivity to cisplatin and paclitaxel, common chemotherapy drugs to treat ovarian cancer111,112,114. miR-18 negatively regulates the tumor-suppressor gene programmed cell death 4 (PDCD4), which regulates the initiation and progression of ovarian cancer114. In contrast, miR-199a increases the response to the same drugs by interfering with the ATP Binding Cassette Subfamily G Member 2 (ABCG2) gene which is associated with multidrug resistance in ovarian cancer-initiating cells114.
Table 4. Examples of how several micro-RNAs (miRNAs) are deregulated, and the downstream effects of that dysregulation in ovarian cancer. This table was adapted and modified from (113).
|
miRNA |
Alteration |
Effect |
Mechanism of deregulation |
|
miR-22 |
Down-regulated |
Tumor suppressor |
− |
|
miR-31 |
Down-regulated |
Tumor suppressor |
CNV |
|
miR-34a/b/c |
Down-regulated |
Tumor suppressor |
Promoter methylation, CNV and p53 mutation |
|
miR-127-3p |
Down-regulated |
Related to drug resistance |
Imprinting, CNV, promoter methylation |
|
miR-181a-3p |
Down-regulated |
− |
CNV, promoter methylation |
|
miR-20a |
Up-regulated |
Oncogenic miRNA |
− |
|
miR-182 |
Up-regulated |
Putative oncogenic miRNA |
CNVs, promoter methylation |
|
miR-200 |
Up-regulated |
Oncogenic miRNA |
CNV |
|
miR-203 |
Up-regulated |
− |
Promoter methylation |
|
miR-205 |
Up-regulated |
Putative oncogenic miRNA |
Promoter methylation |
CNV: Copy number variation; −: not known.
miRNA as Biomarkers
miRNAs can target mRNAs for cleavage or translational repression. They are not capable of coding for proteins but instead possess necessary structural, catalytic, and regulatory functions as they are located at fragile sites and genomic regions related to cancer115. Approximately 30% of the genes that code for human proteins are susceptible to miRNAs. Aberrant changes in miRNA expressions can be linked to the expression of oncogenes or protooncogenes116. miRNAs can be formidable as they can sustain themselves in fixed tissue, blood, and other body fluids which makes them an excellent source of biomarkers. It was seen that miRNA expression profiles were invaluable in detecting and identifying progenitor cells for different cancer types117. miRNAs stored in serum also exhibit astounding resistance to enzymatic cleavage by RNase A, therefore being able to maintain their intrinsic stability for a prolonged period115.
Discovery of novel biomarkers is one of the major challenges in diagnostics. It is known that certain miRNAs are involved in regulating cell proliferation, differentiation, and death111,112,116. The microarray platform has been used to determine 29 differentially expressed miRNAs with ovarian cancer. From the analysis, it was found that miR-141, miR-200a, miR-200b, and miR-200c were up-regulated, whereas, miR-125b1, miR-140, miR-145, and miR-199a were down-regulated118. Kosaka et al. detected elevated expression of miR-21, miR-92, miR-93, miR-126, and miR-29a from serum119. Additionally, the miRNA expression patterns in ovarian tumors also provide useful insights into the histological subtypes of ovarian tumors116. There are multiple ways of detecting miRNAs including in-situ hybridization, northern blotting, microarrays, next-generation sequencing, enzymatic luminescence miRNA assay, and nanopore technology115. In a previously reported study, a patient with ovarian cancer negative for the diagnostic Cancer Antigen 125 (CA-125) threshold did, in fact, have elevated levels of miR-21, miR-92, miR-93, and miR-126. This shows that using miRNA as a biomarker may be more accurate for ovarian cancer diagnosis, especially in non-epithelial-types116,117. Tables 5-7 lists miRNAs that are used as potential biomarkers for diagnosing various types of ovarian cancers.
Ovarian Cancer Stem Cells
Cancer stem cells (CSCs) related to ovarian cancer are considered the emerging culprit in primary tumorigenesis relapse, metastasis and chemoresistance11. CSCs are malignant multi-potent progenitors hypothesized to possess mutations making them drug resistant119. They are suspected to exhibit chemo-resistant properties, including low mitotic index, hypoxemia resistance, enhanced DNA-repair mechanisms, and up-regulation of ATP-dependent membrane transporters that selectively export chemotherapy agents119,120. Studies have demonstrated the emergence of CSCs after chemotherapy treatment with cisplatin and paclitaxel121,122. High levels of Zinc Finger CCHC Domain-Containing Protein (LIN28), an RNA-binding protein, are expressed in ovarian cancer cells123. Cells with high levels of LIN28 are postulated to secrete exosomes which are taken up by HEK293 cells, transforming them into an invasive malignant phenotype123,124. The exosomes resulted in induction of EMT in ovarian cancer cells123,125. Another study found that ovarian cancer stem cell cultures expressed mesenchymal and epithelial markers, including cytoplasmic E-cadherin and cell surface markers CD133 and CD44126. A culture of ovarian clear-cell carcinoma cells with epithelial E-cadherin-positive and mesenchymal vimentin-positive biomarkers demonstrated cooperation between these two phenotypes. These coexist at the epithelial edge of the culture, which suggests a cooperative interaction and the possibility that mesenchymal cells influence epithelial cells to transition127.
Table 5. Types of specific micro-RNA biomarkers in ovarian cancer. The table was slightly modified and adapted from (115).
|
Diagnostic |
let-7 family, miR-21, miR-29a, miR-92, miR-93, miR-126, miR-127, miR-132, miR-144, miR-155, miR-182, miR-200 family, miR-205, miR-214, miR-222, miR-302 |
|
Prognostic |
miR-141, miR-200 family, miR-429, miR-410, miR-645 |
|
Predictive |
let-7g, miR-23a, miR-27a, miR-30c, miR-181a, miR-181b, miR-199a-3p, miR-213 |
Table 6. Micro-RNAs as potential biomarkers in formalin-fixed paraffin-embedded ovarian tumors. The table was slightly modified and adapted from (115).
|
Up-regulated |
miR-223 |
|
Down-regulated |
miR-9, miR-200a, miR-200b, miR-429 |
Table 7. Ovarian cancer micro-RNA (miRNA) biomarkers in blood and other bodily fluids. The table was slightly modified and adapted from (115).
|
Source |
miRNA (Biomarker) |
Condition |
|
Serum |
miR-21, miR-92, miR-93, miR-126, miR-29a |
Up-regulated |
|
miR-155, miR-127, miR-99b |
Down-regulated |
|
|
Plasma |
miR-205 |
Up-regulated |
|
let-7f |
Down-regulated |
|
|
Exosome |
miR-21, miR-141, miR-200a/b/c, miR-203, miR-205, miR-214 |
Up-regulated |
Effects of HDAC inhibitor, DNA Methyltransferase 1 inhibitor and Calpain inhibitor Combination Therapy on Ovarian Cancer Cells
HDAC inhibitors are emerging as an alternative and novel type of anticancer agent. Acetylation of histone stimulates expression of genes that inhibit proliferation while inducing apoptosis of cancer cells128. Deacetylation of histone by deacetylase reduces the expressions of tumor suppressors such as p21WAF1, growth arrest and DNA damage (GADD45) and several others129-131. Hydroxamic acid was reported as potent HDAC inhibitor which interacts with the zinc in deacetylase132. It was seen that HDAC was inhibited in neuroblastoma cell line SH-SY5Y by 2-amino-5-(thiophen-2-yl) benzamide series (K560). The 2-aminobenzamide moiety in K560 exerts selective inhibition by interacting with the zinc binding site within the HDAC1/2 pocket133-134.
DNA methyltransferase 1 (DNMT1) inhibitors can remove methylation resulting in expression Regulator of G protein signaling 10 (RGS10), a regulator of cell survival and chemo resistance. These events increase susceptibility of ovarian cancer cells to cisplatin135. DNMT1 can be broken down by DNMT1 inhibitors resulting in CPG demethylation, causing re-expression of tumor suppressor genes p21, p16, and retinoic acid receptor beta 2 (RARB2)136. DNMT1 inhibitors in tandem with TNF-related apoptosis inducing ligand (TRAIL) inhibitory antibodies were found to reduce breast tumor size in a xenograft mouse model137. DMT1 inhibitor, when combined with telomere inhibitor GT-oligo, caused a significant reduction in ovarian cancer proliferation137,138. Growth of several types of cancer, such as breast, ovarian, prostate, and leukemia, were affected when DNMT1 inhibitors were combined with calpain protease inhibitor139,140. Calpain, a ubiquitous protease, regulates multiple signaling proteins and their pathways and disruption in calpain expression is known to be a pathological event in cancer formation. Lapinska et al. studied the effects of two structurally different DNMT1 inhibitors: Class 1 sodium butyrate, and class 11 sub eranilo-hydroxamic acid (SAHA) both combined with the calpain protease inhibitor, calpeptin. In the study, two distinct ovarian cancer cell lines, CAOV-3 and SKOV3, were used. Results showed significant growth inhibition of both ovarian cancer cell lines140. DNMT1 inhibitors when combined with calpeptin resulted in the inhibition of cell-cycle progression, a decrease in metastatic activity, programmed cell death, autophagy, and re-expression of tumor-suppressor genes such as ADP-ribosylarginine hydrolase (ARH1), p21 and RARB2140. Therefore, combination therapy, when used effectively, can be a potential treatment option for ovarian cancer.
Bromodomain and Extra-terminal Domain (BET) Bromodomain Inhibition
BET bromodomain inhibitors interrupt acetyl lysine recognition by replacing BET bromodomain proteins from transcriptional complexes, resulting in gene inhibition141. BRd4, a member of BET (bromodomain and extra-terminal domain) family, activity is known to be important for tumor progression. Inhibition of BRd4 was found to interfere with transcription of oncogenes, causing tumor regression and apoptosis142,143. Targeting using BET bromodomain inhibitors was shown to affect kinome dynamics in ovarian cancer cell lining144. Baratta et al. found that resistance to BET bromodomain inhibitors involved the downstream signaling pathways of P13K, AKT, and MAPK/ERK alongside the activation of receptor tyrosine kinases and prolonged contact with a BET bromodomain inhibitor JQ1 resulted in accelerated susceptibility of ovarian cancer cells to combination therapies targeting the pathways mentioned above142,143. Unfortunately, combination therapies involving inhibitors such as BET bromodomain inhibitors attack kinases that are involved in other critical and beneficial pathways142,144. BET bromodomain inhibitor-mediated apoptosis by JQ1 showed cell-specific effects on different ovarian cancer cell lines144. Gene-expression studies using RNA sequencing in A1847 and OVCAR5 ovarian cancer cells following JQ1 therapy were performed to see how it affected RNA expression of BRd4 targets145. According to the results, RNA expression was reduced for Interleukin-7 receptor (IL7R), FOXM1, CDK4, CDK6, MYC and FOS-like antigen (FOSL), which were established to be BRd4 targets145. The majority of studies concluded that BET bromodomain inhibitor therapies may not be suitable to provide long-term treatment benefits due to acquired drug resistance of ovarian cancer146.
Epigenetic Therapies to Overcome Chemotherapy Resistance
Epigenetic therapies largely target histone deacetylation or DNA methylation as means of reversing these damaging epigenetic DNA changes147. Hypermethylation of CpG-rich promoter regions can result in gene silencing, and when tumor-suppressor genes are silenced, destabilization of the cell cycle may occur. More specifically, these epigenetic changes can prevent cell apoptosis and permit uncontrolled proliferation, leading to the formation of a tumor147. Histone deacetylation is also damaging, as it can destabilize tumor-suppressor genes and result in tumor formation. The two drugs which focus on these epigenetic issues are DNMTs and HDAC inhibitors148. Over the past few decades, researchers have focused on these epigenetic therapies as a means of treating multiple cancer types147,148. Hydralazine, an oral antihypertensive and non-nucleoside DNMT inhibitor, inhibits DNMT1 activity by reducing DNMT1 and DNMT3A expression148. In cervical cancer, hydralazine demethylation and gene-reactivating activity were observed149. Valproic acid, an anti-epileptic fatty acid, resulted in N-terminal hyperacetylation of H3 and H4, resulting in blocking substrate access in ovarian cancer cells150.
Complementary and Alternative Interventions and Targeting Epigenetic Pathways
Human beings have traditionally utilized medicinal plants against cancer worldwide. Lawsonia intermis (henna) is used as an agent for blood cancer in Ayurveda and Unani medicine, and used as anticancer drug in Sudan151,152. Trigonella foenum-graecum (fenugreek) is traditionally utilized to treat high cholesterol, diabetes, wound inflammation, and cancer153-155. Ambrosa maritima, a Sunanese traditional medicine, is widely used for several kinds of diseases including malignancies156. Compounds in plants have been reported to inhibit migration, invasion, and proliferation of cancer cells. Indeed, many dietary components are linked to affecting DNA methylation or regulating histone modifications such as isothiocyanates, genistein, resveratrol, epigallocatechin-3-gallate and quercetin. Many studies were performed to examine these types of compounds for preventing and treating cancer. Quercetin a representative flavonoid, ameliorates ROS-mediated cellular damage, and prevents tumor invasion and metastasis by reducing expression of MMP2 and -9157,158. Quercetin-3-O-glucuronide also inhibits MMP2 and 9 expression and cell invasion via suppressing beta 2 adrenergic receptor (β2-AR) signaling in noradrenaline-stimulated breast cancer cells159.
DNA methylation is catalyzed by DNA methyltransferase, which is known to silence tumor-suppressing genes. DNA methyltransferase has been an attractive target to epigenetically prevent or treat malignancies160. Cytosine analogs such as 5-azacytidine and 5-aza-2'-deoxycytidine can act as demethylating agents by replacing cytosines in DNA thereby inhibiting DNA methyltransferase161,162. Some efficacy has been reported for the use of hypomethylating agents in blood cancer such as myelodysplastic syndromes but there are fewer data for solid tumors. There is some evidence that treatment with decitabine may reinvigorate the sensitization of platinum-resistant ovarian tumors163. 2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)-3-(1H-indol-3-yl) propanoic acid (RG108) was identified and used as a different type of potent inhibitor of DNA methyltransferase but has not been tested for ovarian cancer. RG108 suppresses DNA transferase via combining with its active moiety163,164.
Studies on pharmacological interventions against ovarian cancer have also been reported. Genistein, 4',5,7-trihydroxyisoflavone, a type of flavonoid found in soybeans, peas, lentils, and other beans165, was reported to inhibit EMT in ovarian cancer cells. Apicidin, a fungal metabolite, is a cyclic tetrapeptide that inhibits migration and proliferation of SKOV-3 cell by down-regulating HDAC 4 and MMP2166. Guadecitabine (SGI-110) acts as a demethylating agent and up-regulates tumor-suppressor genes167. Advanced stages of ovarian cancer can negate the effects of certain inhibitors of demethylating agents, such as fazarabine168.
Alternative Cancer Therapies
Alternative forms of chemotherapy are becoming progressively popular in cancer treatment. The health hazards of radiation therapy and increasing tolerance of cancer cells to platinum-based drugs has geared research towards different phytochemicals and novel drug delivery methods169. Afroze et al. studied the differential effects of a cardiotonic steroid, cinobufotalin on three different ovarian cancer cells and found that cinobufotalin affected the growth and metastatic capacity of ovarian cancer cells170. Some common natural compounds that are being explored to investigate their anticancer properties are flavonoids, garlic, turmeric, stinging nettle, green tea, and many more. Yamauchi et al. investigated the effect of a modified quercetin derivative known as 3’4’7-O-trimetheylquercetin (347TMQ) which inhibited the invasion and migration of three types of ovarian cancer cell171. Ashraf et al. followed up on this investigation and determined that 347TMQ resulted in expression of pro-apoptotic proteins causing apoptosis in three different ovarian cancer cell lines172. Therapies using these compounds address some of the pervasive difficulties associated with cancer diagnosis, treatment, and survival169,173.
Cancer cells take advantage of the Warburg effect whereby glycolysis is upregulated resulting in an increased lactate accumulation174. In cancer cells, NADH that is produced by glyceraldehyde phosphate dehydrogenase (GAPDH) and must be used up to regenerate nicotinamide adenine dinucleotide (NAD+) to continue glycolysis. This high rate of glycolysis supports a vigorous metabolic environment that allows for the rapid growth of cancer cells175. Coverdale et al. was able to increase the lipophilicity of cancer cells by delivering intact active organo-osmium catalysts inside cells. This made cancer cells more susceptible to metallodrugs therefore, maximizing anticancer effects176. Coverdale et al. used a synthesized osmium molecule in tandem with sodium formate to manipulate pyruvate breakdown in cancer cells, resulting in decreased proliferation and increased apoptosis174. The combination of the sodium formate and osmium reduced NAD+ to NADH inside cancer cells using non-chiral osmium catalysts176. This novel approach targets the inherent redox vulnerability of cancer cells, which results from dysfunctional mitochondria174. A study by Wargovich et al. showed that phase I and phase II metabolic processes by natural compounds may have similar anticancer activity177. Using osmium complex with higher concentrations of sodium formate significantly reduced the proliferation of A2780 cancer cells by causing G1 cell-cycle arrest. This mechanism of action prevents DNA damage by preventing nuclear accumulation of osmium and causing S/G2M cell cycle arrest174,178.
Thus, many of the epigenetic foci related to the pathogenesis of ovarian cancer have been reviewed along with types of epigenetic modifications that have been elicited in the available research literature. In addition, the role of miRNA and cancer stem cells are discussed in this growing area of research related to oncogenic pathways related to ovarian neoplasms. This is followed by a summary of research regarding the targeting of epigenetic pathways with alternative and combination related therapies. Given that ovarian cancer is still a leading cause of cancer-related death and is commonly diagnosed at latter stages and with continued issues of treatment resistance, more research regarding the underlying mechanisms and possible treatment modalities are warranted.
Acknowledgements
Funding of this study was supported by The Discovery Foundation, (MNU) and Grimes Family Foundation Endowment in Gynecologic Oncology (MNU). All authors have read the Journal’s authorship agreement.
Conflicts of Interest
The Authors have expressed no conflicts of interest.
Authors’ Contributions
Composing and preparing the article: A. H. M. Zuberi Ashraf, Syeda H. Afroze, Grace A. Osuji, Saba Y. Kayani and Natalie Colon
Reviewing the article: Thomas J. Kuehl and Kimberly A. Pilkinton.
Planning, reviewing, and approving of the article for submission: Mohammad N. Uddin.
References
- Cancer Stat Facts: Ovarian Cancer. National Cancer Institute, 2015 Available at: http://seer.cancer.gov/statfacts/html/ovary.html. Last accessed on 9th June 2019.
- Kumar V, Abbas AK, Fausto N, et al Robbins and Cotran Pathologic Basis of Disease. Ninth (eds.). Philadelphia PA Elsevier Health Sciences. 2014; 991-1042.
- About Ovarian Cancer: What is Ovarian Cancer? American Cancer Society, 2015. Available at: http://www.cancer.org/cancer/ovariancancer/detailedguide/ovarian-cancer-what-is-ovarian-cancer. Last accessed on 9th June 2019.
- Kotsopoulos J, Lubinski J, Gronwald J, et al Factors influencing ovulation and the risk of ovarian cancer in BRCA1 and BRCA2 mutation carriers. Hereditary Breast Cancer Clinical Study Group. Int J Cancer. 2015; 137(5): 1136-1146. PMID: 25482078. DOI: 10.1002/ijc.29386
- Reade CJ, Riva JJ, Busse JW, et al. Risks and benefits of screening asymptomatic women for ovarian cancer: a systematic review and meta-analysis. Gynecol Oncol. 2013; 130: 674-681. PMID: 23822892. DOI: 10.1016/j.ygyno.2013.06.029
- Karst AM, Drapkin R: Ovarian cancer pathogenesis: A model in evolution. J Oncol. 2010; 2010: 1-13. PMID: 24265397. DOI:10.1155/2010/932371
- Kurman RJ. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann Oncol. 2013; 24(suppl_10): 16-21. PMID: 24265397. DOI: 10.1093/annonc/mdt463
- Nezhat FR, Apostol R, Nezhat C, et al. New insights in the pathophysiology of ovarian cancer and implications for screening and prevention. Am J Obstet Gynecol. 2015; 213(3): 262-267. PMID: 25818671. DOI: 10.1016/j.ajog.2015.03.044
- Kurman RJ, Shih IM: The Origin and pathogenesis of EOC-a proposed unifying theory. Am J Surg Pathol. 2010; 34(3): 433-443. PMID: 20154587. DOI: 10.1097/PAS.0b013e3181cf3d79
- Kurman RJ, Shih IM: Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm. Hum Pathol. 2011; 42(7): 918-931. PMID: 21683865. DOI: 10.1016/j.humpath.2011.03.003
- Wang Y, Mang M, Wang Y, et al. Tubal origin of ovarian endometriosis and clear cell and endometrioid carcinoma. Am J Cancer Res. 2015; 5(3): 869-879. PMID: 26045974. DOI: ajcr0004931
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008; 358(11): 1148–1159.
- Dominguez M. Interplay between Notch signaling and epigenetic silencers in cancer. Cancer Res. 2006; 66(18): 8931-8934. PMID: 16982729. DOI: 10.1158/0008-5472.CAN-06-1858
- Aithal MG, Rajeswari N. Role of Notch signaling pathway in cancer and its association with DNA methylation. J Genet. 2013; 92(3): 667-675. PMID: 24371188
- Rose SL. Notch Signaling Pathway in ovarian cancer. Int J Gynecol Cancer. 2009; 19(4): 564-566. PMID: 19509550. DOI: 10.1111/IGC.0b013e3181a12ed2
- Lu KH, Patterson AP, Wang L, et al. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res. 2004; 10(10): 3291-3300. PMID: 15161682. DOI: 10.1158/1078-0432.CCR-03-0409
- Jung SG, Kwon YD, Song JA, et al.: Prognostic significance of Notch 3 gene expression in ovarian serous carcinoma. Cancer Sci. 2010; 101(9): 1977-1983. PMID: 20624166. DOI: 10.1111/j.1349-7006.2010.01641.x
- Korver W, Rouse J, Heinen K, et al. The human trident/HFH-11/FKHL 16 gene: Structure/localization, and promoter characterization. Genomics. 1997; 6(3): 435-442. PMID: 9441747. DOI: 10.1006/geno.1997.5065
- Leung TW, Lin SS, Tsang AC, et al. Overexpression of FOXM1 stimulates cyclin B1 expression. FEBS Lett. 2001; 507(1): 59-66. PMID: 11682060. DOI: 10.1016/S0014-5793(01)02915-5
- Tan Y, Raychaudhuri P, Costa RH. CHK2 mediates stabilization of the FOXM1 transcription factor to stimulate expression of DNA repair genes. Mol Cell Biol. 2007; 27(3): 1007-1016. PMID: 17101782. DOI: 10.1128/MCB.01068-06
- Jaiswal N, Chakraborty S, Nag A. Biology of FOXM1 and its emerging role in cancer therapy. Journal of Proteins and Proteomics. 2014; 5(1): 1-24.
- Hwang S, Mahadevan S, Qadir F, et al. Identification of FOXM1âinduced epigenetic markers for head and neck squamous cell carcinomas. Cancer. 2013; 119(24): 4249-4258. PMID: 24114764. DOI: 10.1002/cncr.28354
- Pilarsky C, Wenzig M, Specht T, et al. Identification and Validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004; 6(6): 744-750. PMID: 15720800. DOI: 10.1593/neo.04277
- Xie Z, Jan G, Ding M, et al. FOXM1 transcription factor is required for maintenance of pluripotency of p19 embryonal carcinoma cells. Nucleic Acid Res. 2010; 38(22): 8027-8038. PMID: 20702419. DOI: 10.1093/nar/gkq715
- Gemenetzidis E, Elena Costea D, Parkinson EK, et al. Induction of human epithelial stem/progenitor expansion by FOXM1. Cancer Res. 2010; 70(22): 9515-9526. PMID: 21062979. DOI: 10.1158/0008-5472.CAN-10-2173
- Ghosh Choudhury T, Loomans HA, Wan YW, et al. Hyperactivation of FOXM1 drives ovarian cancer. Growth and metastasis independent of the G2 to M cell cycle checkpoint. Cancer Res. 2013; 73(8 supp): Abstract 3113. DOI: 10.1158/1538-7445.AM2013-3113
- Lok GT, Chan DW, Liu VW, et al. Aberrant activation of ERK/FOXM1 signaling cascade triggers the cell migration/invasion in ovarian cancer cells. PLOS ONE. 2011; 6(8): 1-10. PMID: 21858223. DOI: 10.1371/journal.pone.0023790
- Gemenetzidis E, Bose A, Riaz AM, et al. FOXM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation. PLOS ONE. 2009; 4(3): 1-18. PMID: 19287496. DOI: 10.1371/journal.pone.0004849
- Wodarz A, Nusse R. Mechanisms of WNT signaling in development. Annu Rev Cell Dev Biol. 1998; 14(1): 59-88. PMID: 9891778. DOI: 10.1146/annurev.cellbio.14.1.59
- Su HY, Lai HC, Lin YW, et al. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through WNT signaling pathway. Int J Cancer. 2010; 127(3): 555-567. PMID: 19957335. DOI: 10.1002/ijc.25083
- Zhu J, Zhang S, Gu L, et al. Epigenetic silencing of DKK2 and WNT signal pathway components in human ovarian carcinoma. Carcinogenesis. 2012; 33(12): 2334-2343. PMID: 22964660. DOI: 10.1093/carcin/bgs278
- Mao B, Niehrs C. Kremen2 modulates Dickkopf2 activity during WNT/LRP6 signaling. Gene. 2003; 302(1): 179-183. PMID: 12527209. DOI: 10.1016/S0378-1119(02)01106-X
- Rask K, Nilsson A, Brännström M, et al. WNT-signalling pathway in ovarian epithelial tumors: increased expression of β-catenin and GSK3β. Br J Cancer. 2003; 89(7): 1298-1304. PMID: 14520463. DOI: 10.1038/sj.bjc.6601265
- Fang D, Chen H, Zhu JY, et al. Epithelial–mesenchymal transition of ovarian cancer cells is sustained by Rac1 through simultaneous activation of MEK1/2 and Src signaling pathways. Oncogene. 2017; 36(11): 1546-1558. PMID: 27617576. DOI: 10.1038/onc.2016.323
- Takai M, Terai Y, Kawaguchi H, et al. The EMT (epithelial-mesenchymal-transition)-related protein expression indicates the metastatic status and prognosis in patients with ovarian cancer. J Ovarian Res. 2014; 7(76): 1-8. PMID: 25296567. DOI: 10.1186/1757-2215-7-76
- Yun-Ju Huang R, Yee Chung V, Paul Thiery J. Targeting pathways contributing to epithelial-mesenchymal transition (EMT) in EOC. Curr Drug Targets. 2012; 13(13): 1649-1653. PMID: 23061545. DOI: 10.2174/138945012803530044
- Kwon MJ, Shin YK. Epigenetic regulation of cancer-associated genes in ovarian cancer. Int J Mol Sci. 2011; 12: 983-1008. DOI:10.3390/ijms12020983
- Iacobuzio-Donahue CA. Epigenetic changes in cancer. Annu Rev Pathol. 2009; 4: 229–249. DOI: 10.1146/annurev.pathol.3.121806.151442
- Schondorf T, Ebert MP, Hoffmann J, et al. Hypermethylation of the PTEN gene in ovarian cancer cell lines. Cancer Lett. 2004; 207(2): 215–220. PMID: 15072831. DOI: 10.1016/j.canlet.2003.10.028
- Baldwin RL, Nemeth E, Tran H, et al. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. 2000; 60(19): 5329–5333. PMID: 11034065.
- Feng W, Marquez RT, Lu Z, et al. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer. 2008; 112(7): 1489–1502. PMID: 18286529. DOI: 10.1002/cncr.23323
- Ibanez de Caceres C, Esteller M, Herman JG, et al. Tumor cell specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 2004; 64(18): 6476–6481. PMID: 15374957. DOI: 10.1158/0008-5472.CAN-04-1529 â¨
- Zhang H, Zhang S, Cui J, et al. Expression and promoter methylation status of mismatch repair gene hMLH1 and hMSH2 in EOC. Aust N Z J Obstet Gynaecol. 2008; 48(5): 505–509. PMID: 19032668. DOI: 10.1111/j.1479-828X.2008.00892.x
- Meng CF, Dai DQ, Guo KJ. Effects of 5-aza-2′-deoxycytidine and trichostatin A on DNA methylation and expression of hMLH1 in ovarian cancer cell line COC1/DDP. Ai Zheng. 2008; 27(12): 1251–1255. PMID: 19079988.
- Cai J, Xu L, Tang H, et al. The role of the PTEN/PI3K/AKT pathway on prognosis in EOC: A meta-analysis. Oncologist. 2014; 19(5): 528-535. PMID: 24718516. DOI: 10.1634/theoncologist.2013-0333
- Sui H, Fan ZZ, Li Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/P-Gp-mediated multiple drug resistance in human cancer cells. J Int Med Res. 2012; 40(2): 426-435. PMID: 22613403. DOI: 10.1177/147323001204000204
- Hennessy BT, Timms KM, Carey MS, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010; 28(22): 3570-3576. PMID: 20606085. DOI: 10.1200/JCO.2009.27.2997
- Chan KY, Ozçelik H, Cheung AN, et al. Epigenetic factors controlling the BRCA1 and BRCA2 genes in sporadic ovarian cancer. Cancer Res. 2002; 62(14): 4151-4156. PMID: 12124354.
- Wu J, Lu Ly, Yu X. The role for BRCA1 in DNA damage response. Protein Cell. 2010; 1(2): 117-123. PMID: 21203981. DOI: 10.1007/s13238-010-0010-5
- Patel KJ, Veronica PC, Lee H, et al. Involvement of BRCA2 in DNA repair. Molecular cell. 1998; 1(3): 347-357. DOI: 10.1016/S1097-2765(00)80035-0
- Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol. 2009; 4: 461-487. PMID: 18954285. DOI: 10.1146/annurev.pathol.3.121806.151422
- Kobel M, Huntsman D, Gilks CB. Critical molecular abnormalities in high-grade serous carcinoma of the ovary. Expert Rev Mol Med. 2008; 10: e22. PMID: 18671886. DOI: 10.1017/S146239940800077X
- Pradhan M, Risberg BA, Tropé CG, et al. Gross genomic alterations and gene expression profiles of high-grade serous carcinoma of the ovary with and without BRCA1 inactivation. BMC cancer. 2010; 10(1): 493-501. PMID: 20843305. DOI: 10.1186/1471-2407-10-493
- Gau DM, Lesnock JL, Hood BL, et al. BRCA1 deficiency in ovarian cancer is associated with alteration in expression of several key regulators of cell motility–A proteomics study. Cell Cycle. 2015; 14(12): 1884-1892. PMID: 25927284. DOI: 10.1080/15384101.2015.1036203
- Shi H, Li Y, Wang X, et al. Association between RASSF1A promoter methylation and ovarian cancer: a meta-analysis. PLOS ONE. 2013; 8(10): 1-7. PMID: 24116157. DOI: 10.1371/journal.pone.0076787
- Barton CA, Hacker NF, Clark SJ, et al. DNA methylation changes in ovarian cancer: implications for early diagnosis, prognosis and treatment. Gynecol. Oncol. 2008; 109(1): 129-139. PMID: 18234305 DOI: 10.1016/j.ygyno.2007.12.017
- Hesson LB, Cooper WN, Latif F. The role of RASSF1A methylation in cancer. Dis Markers. 2007; 23(1-2): 73-87. PMID: 17325427. DOI: 10.1155/2007/291538
- Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci. 2007; 120(18): 3163-3172. DOI:10.1242/jcs.010389
- Pfeifer GP, Dammann R. Methylation of the tumor suppressor gene RASSF1A in human tumors. Biochem (Mosc). 2005; 70(5): 576-583. PMID: 15948711. l
- Feinberg AP. Cancer epigenetics takes center stage. Proc Natl Acad Sci USA. 2001; 98(2): 392-394. PMID: 11209042. DOI: 10.1073/pnas.98.2.392
- Peng H, Xu F, Pershad R, et al. ARHI is the center of allelic deletion on chromosome 1p31 in ovarian and breast cancers. Int J Cancer. 2000; 86(5): 690-694. PMID: 10797292. DOI: 10.1002/(SICI)1097-0215(20000601)86:5<690::AID-IJC14>3.0.CO;2-K
- Johnson MD, Wu X, Aithmitti N, et al.: PEG3/PW1 is a mediator between p53 and BAX in DNA damage-induced neuronal death J Biol Chem. 2002; 277(25): 23000-23007. PMID: 11943780. DOI: 10.1074/jbc.M201907200
- Kohler MF, Marks JR, Wiseman RW, et al. Spectrum of mutation and frequency of allelic deletion of the p53 gene in ovarian cancer. J Natl Cancer Inst Monogr. 1993; 85(18): 1513-1519. PMID: 8360934. DOI: 10.1093/jnci/85.18.1513
- Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993; 260(5109): 816-819. PMID: 8484122. DOI: 10.1126/science.8484122
- Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH 1 is associated with hereditary non-polyposis colon cancer. Nature. 1994; 368(6468): 258-261. PMID: 8145827. DOI: 10.1038/368258a0
- Murphy MA, Wentzensen N. Frequency of mismatch repair deficiency in ovarian cancer: a systematic review. Int J Cancer. 2011; 129(8): 1914-1922. PMID: 21140452. DOI: 10.1002/ijc.25835
- Geisler JP, Goodheart MJ, Sood AK, et al. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer. 2003; 98(10): 2199-2206. PMID: 14601090. DOI: 10.1002/cncr.11770
- Aebi S, Kurdi-Haidar B, Gordon R, et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996; 56(13): 3087-3090. PMID: 8674066
- Wang J, Yu L, Cai J, et al. The role of EZH2 and DNA methylation in hMLH1 silencing in EOC. Biochem Biophys Res Commun. 2013; 433(4): 470-476. PMID: 23523787. DOI: 10.1016/j.bbrc.2013.03.037
- Seeber LMS, Van Diest PJ. Epigenetics in ovarian cancer. Methods Mol Biol. 2012; 863: 253-269. DOI 10.1007/978-1-61779-612-8_15
- Su HY, Lai HC, Lin YW, et al. An epigenetic marker panel for screening and prognostic prediction of ovarian cancer. Int J Cancer. 2009; 124(2): 387–393. PMID: 18942711. DOI: 10.1002/ijc.23957
- Strathdee G, Vass JK, Oien KA, et al. Demethylation of the MCJ gene in stage III/ IV EOC and response to chemotherapy. Gynecol Oncol. 2005; 97(3): 898–903. PMID: 15894365. DOI: 10.1016/j.ygyno.2005.03.023
- Czekierdowski A, Czekierdowska S, Wielgos M, et al. The role of CpG islands hypomethylation and abnormal expression of neuronal protein synuclein-gamma (SNCG) in ovarian cancer. Neuro Endocrinol Lett. 2006; 27(3): 381–386. PMID: 16816827
- Gupta A, Godwin AK, Vanderveer L, et al. Hypomethylation of the synuclein γ gene CpG island promotes its aberrant expression in breast carcinoma and ovarian carcinoma. Cancer Res. 2003; 63(3): 664-673. PMID: 12566312.
- Yao X, Hu JF, Li T, et al. Epigenetic regulation of the taxol resistance–associated gene TRAG-3 in human tumors. Cancer Genet Cytogenet. 2004; 151(1): 1-3. PMID: 15120907. DOI: 10.1016/j.cancergencyto.2003.08.021
- Woloszynska-Read A, James SR, Link PA, et al. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun. 2007; 7(1): 21-31. PMID: 18095639
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001; 409(6822): 860-921. PMID: 11237011. DOI: 10.1038/35057062
- Earp MA, Cunningham JM. DNA methylation changes in EOC histotypes. Genomics. 2015; 106(6): 311-321. PMID: 26363302. DOI: 10.1016/j.ygeno.2015.09.001
- Sung HY, Yang SD, Park AK, et al.Aberrant hypomethylation of solute carrier family 6-member 12 promoter induces metastasis of ovarian cancer. Yonsei Med J. 2017; 58(1): 27-34. PMID: 27873492. DOI: 10.3349/ymj.2017.58.1.27
- Zhou Y, Holmseth S, Hua R, et al. The betaine-GABA transporter (BGT1, SLC6A12) is predominantly expressed in the liver and at lower levels in the kidneys and at the brain surface. Am J Physiol Renal Physiol. 2012; 302(3): 316-328. PMID: 22071246. DOI: 10.1152/ajprenal.00464.2011
- Sung HY, Ju W, Ahn JH. DNA hypomethylation-mediated overexpression of carbonic anhydrase 9 induces an aggressive phenotype in ovarian cancer cells. Yonsei Med J. 2014; 55(6): 1656-1663. PMID: 25323905. DOI: 10.3349/ymj.2014.55.6.1656
- De Leon M, Cardenas H, Vieth E, et al. Transmembrane protein 88 (TMEM88) promoter hypomethylation is associated with platinum resistance in ovarian cancer. Gynecol Oncol. 2016; 142(3): 539-547. PMID: 27374141. DOI: 10.1016/j.ygyno.2016.06.017
- Hill RE, Jones PF, Rees AR, et al. A new family of mouse homeo box-containing genes: molecular structure, chromosomal location, and developmental expression of Hox-7.1. Genes Dev. 1989; 3(1): 26-37. PMID: 2565278. DOI: 10.1101/gad.3.1.26
- Bonito NA, Borley J, Wilhelm-Benartzi CS, et al. Epigenetic regulation of the homeobox gene MSX1 associates with platinum-resistant disease in high-grade serous EOC. Clin Cancer Res. 2016; 22(12): 3097-3104. PMID: 26763252. DOI: 10.1158/1078-0432.CCR-15-1669
- Oehler VG, Guthrie KA, Cummings CL, et al. The preferentially expressed antigen in melanoma (PRAME) inhibits myeloid differentiation in normal hematopoietic and leukemic progenitor cells. Blood. 2009; 114(15): 3299-3308. PMID: 19625708. DOI: 10.1182/blood-2008-07-170282
- Zhang W, Barger CJ, Link PA, et al. DNA hypomethylation-mediated activation of cancer/testis antigen 45 (CT45) genes is associated with disease progression and reduced survival in EOC. Epigenetics. 2015; 10(8): 736-748. PMID: 26098711. DOI: 10.1080/15592294.2015.1062206
- Kanwal R, Gupta S. Epigenetic modifications in cancer. Clinical genetics. 2012; 81(4): 303-311. PMID: 22082348. DOI: 10.1111/j.1399-0004.2011.01809.x
- Marsh DJ, Shah JS, Cole AJ. Histones and their modifications in ovarian cancer–drivers of disease and therapeutic targets. Front Oncol. 2014; 4(144): 1-12. PMID: 24971229. DOI: 10.3389/fonc.2014.00144
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007; 8(4): 286-298. PMID: 17339880. DOI: 10.1038/nrg2005
- Jang KY, Kim KS, Hwang SH, et al. Expression and prognostic significance of SIRT1 in ovarian epithelial tumors. Pathology. 2009; 41(4): 366-371. PMID: 19404850. DOI: 10.1080/00313020902884451
- Caslini C, Capo-Chichi CD, Roland IH, et al. Histone modifications silence the GATA transcription factor genes in ovarian cancer. Oncogene. 2006; 25(39): 5446–5461. PMID: 16607277. DOI: 10.1038/sj.onc.1209533
- Bracken AP, Dietrich N, Pasini D, et al. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006; 20(9): 1123-1136. PMID: 16618801. DOI: 10.1101/gad.381706
- Ketel CS, Andersen EF, Vargas ML, et al. Subunit contributions to histone methyltransferase activities of fly and worm polycomb group complexes. Mol Cell Biol. 2005; 25(16): 6857-6868. PMID: 16055700 DOI: 10.1128/MCB.25.16.6857-6868.2005
- Li H, Bitler BG, Vathipadiekal V, et al. ALDH1A1 is a novel EZH2 target gene in EOC identified by genome-wide approaches. Cancer Prev Res. 2012; 5(3): 484-491. PMID: 22144423. DOI: 10.1158/1940-6207.CAPR-11-0414
- Li H, Cai Q, Wu H, et al. SUZ12 promotes human EOC by suppressing apoptosis via silencing HRK. Mol Cancer Res. 2012; 10(11): 1462-1472. PMID: 22964433. DOI: 10.1158/1541-7786.MCR-12-0335
- Lu C, Han HD, Mangala LS, et al. Regulation of tumor angiogenesis by EZH2. Cancer cell. 2010; 18(2): 185-197. DOI: 10.1016/j.ccr.2010.06.016
- Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013; 154(3): 490-503. PMID: 23911317. DOI: 10.1016/j.cell.2013.07.011
- Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013; 45(6): 592-601. PMID: 23644491. DOI: 10.1038/ng.2628
- Wei D, Weissman BE. Genetics and Genomics of Malignant Rhabdoid Tumours. eLS. 2014. DOI:10.1002/9780470015902.a0025012
- Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. 2014; 166(3): 350-366. PMID: 25169151. DOI: 10.1002/ajmg.c.31410
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodelers and cancer. Nat Rev Cancer. 2011; 11(7): 481-492. PMID: 21654818. DOI: 10.1038/nrc3068
- Kuwahara Y, Wei D, Durand J, et al. SNF5 reexpression in malignant rhabdoid tumors regulates transcription of target genes by recruitment of SWI/SNF complexes and RNAPII to the transcription start site of their promoters. Mol Cancer Res. 2013; 11(3): 251-260. PMID: 23364536. DOI: 10.1158/1541-7786.MCR-12-0390
- Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv. 2015; 1(5): 1-17. PMID: 26601204. DOI: 10.1126/sciadv.1500447
- Jones S, Wang TL, Shih IM, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010; 330(6001): 228-231. PMID: 20826764. DOI: 10.1126/science.1196333
- Lowery WJ, Schildkraut JM, Akushevich L, et al. Loss of ARID1A-associated protein expression is a frequent event in clear cell and endometrioid ovarian cancers. Int J Gynecol Cancer. 2012; 22(1): 9-14. PMID: 22193641. DOI: 10.1097/IGC.0b013e318231f140
- Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363(16): 1532-1543. PMID: 20942669. DOI: 10.1056/NEJMoa1008433
- Worley MJ, Welch WR, Berkowitz RS, et al. Endometriosis-associated ovarian cancer: a review of pathogenesis. Int J Mol Sci. 2013; 14(3): 5367-5379. PMID: 23466883. DOI: 10.3390/ijms14035367
- Karnezis AN, Wang Y, Ramos P, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcemic type. J Pathol. 2016; 238(3): 389-400. PMID: 26356327. DOI: 10.1002/path.4633
- Jelinic P, Mueller JJ, Olvera N, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat genet. 2014; 46(5): 424-426. PMID: 24658004. DOI: 10.1038/ng.2922
- Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat genet. 2014; 46(5): 427-429. PMID: 24658001 DOI: 10.1038/ng.2928
- Liu T, Qin W, Hou L, et al. MicroRNA-17 promotes normal ovarian cancer cells to cancer stem cells development via suppression of the LKB1-p53-p21/WAF1 pathway. Tumor Biol. 2015; 36(3): 1881-1893. PMID: 25510663. DOI: 10.1007/s13277-014-2790-3
- Kinose Y, Sawada K, Nakamura K, et al.The role of microRNAs in ovarian cancer. Biomed Res Int. 2014; 2014: 1-11. DOI: 10.1155/2014/249393
- Zhang S, Lu Z, Unruh AK, et al. Clinically relevant microRNAs in ovarian cancer. Mol Cancer Res. 2015; 13(3): 393-401. PMID: 25304686. DOI: 10.1158/1541-7786.MCR-14-0424
- Katz B, Tropé CG, Reich R, et al.: MicroRNAs in Ovarian Cancer. Hum Pathol. 2015; 46(9): 1245-1256. PMID: 26216350. DOI: 10.1016/j.humpath.2015.06.013
- Lan H, Lu H, Wang X, et al. MicroRNAs as potential biomarkers in cancer: opportunities and challenges. Biomed Res Int. 2015; 2015: 1-17. PMID: 25874201. DOI: 10.1155/2015/125094
- Zhang B, Cai FF, Zhong XY. An overview of biomarkers for the ovarian cancer diagnosis. Eur J Obstet Gynecol Reprod Biol. 2011; 158(2): 119-123. PMID: 21632171. DOI: 10.1016/j.ejogrb.2011.04.023
- Rosenfeld N, Aharonov R, Meiri E, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008; 26(4): 462-469. PMID: 18362881. DOI: 10.1038/nbt1392
- Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer sci. 2010; 101(10): 2087-2092. PMID: 20624164. DOI: 10.1111/j.1349-7006.2010. 01650.x
- Zhang S, Balch C, Chan MW, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008; 68(11): 4311-4320. PMID: 18519691. DOI: 10.1158/0008-5472.CAN-08-0364
- Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010; 141(1): 69-80. PMID: 20371346. DOI: 10.1016/j.cell.2010.02.027
- Rizzo S, Hersey JM, Mellor P, et al. Ovarian cancer stem cell–like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther. 2011; 10(2): 325-335. PMID: 21216927. DOI: 10.1158/1535-7163.MCT-10-0788
- Kobayashi Y, Seino KI, Hosonuma S, et al. Side population is increased in paclitaxel-resistant ovarian cancer cell lines regardless of resistance to cisplatin. Gynecol Oncol. 2011; 121(2): 390-394. PMID: 21272926. DOI: 10.1016/j.ygyno.2010.12.366
- Yang X, Lin X, Zhong X, et al. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1–positive cancer stem cells. Cancer Res. 2010; 70(22): 9463-9472. PMID: 21045151. DOI: 10.1158/0008-5472.CAN-10-2388
- Peng S, Maihle NJ, Huang Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene. 2010; 29(14): 2153-2159. PMID: 20101213. DOI: 10.1038/onc.2009.500
- Enriquez VA, Cleys ER, Da Silveira JC, et al. High LIN28A-expressing ovarian cancer cells secrete exosomes that induce invasion and migration in HEK293 cells. Biomed Res Int. 2015; 2015: 1-13. PMID: 26583126. DOI: 10.1155/2015/701390
- Strauss R, Li ZY, Liu Y, et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLOS ONE. 2011; 6(1): 1-20. PMID: 21264259. DOI: 10.1371/journal.pone.0016186
- Goyeneche AA, Koch M, Bell MC, et al. Long-term primary culture of a clear cell ovarian carcinoma reveals an epithelial–mesenchymal cooperative interaction. Cancer Cell Int. 2015; 15(88): 1-9. PMID: 26405433. DOI: 10.1186/s12935-015-0243-8
- Liu T, Kuljaca S, Tee A, et al.: Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006; 32(3): 157-165. PMID: 16516391. DOI: 10.1016/j.ctrv.2005.12.006
- Chen Z, Clark S, Birkeland M, et al. Induction and superinduction of growth arrest and DNA damage gene 45 (GADD45) α and β messenger RNAs by histone deacetylase inhibitors trichostatin A (TSA) and butyrate in SW620 human colon carcinoma cells. Cancer let. 2002; 188(1-2): 127-140. PMID: 12406558.
- Burgess AJ, Pavey S, Warrener R, et al. Up-regulation of p21WAF1/CIP1 by histone deacetylase inhibitors reduces their cytotoxicity. Mol Pharmacol. 2001; 60(4): 828-837. PMID: 11562446
- Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell. 2003; 4(1): 13-18. PMID: 12892709. DOI: 10.1016/S1535-6108(03)00165-X
- Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol. 9(2002): 3-16. PMID: 11841934 DOI: https://doi.org/10.1016/S1535-6108(03)00165-X
- Hirata Y, Hirata M, Kawaratani Y, et al. Anti-tumor activity of new orally bioavailable 2-amino-5-(thiophen-2-yl) benzamide-series histone deacetylase inhibitors, possessing an aqueous soluble functional group as a surface recognition domain. Bioorg Med Chem Lett. 2012; 22(5): 1926-1930. PMID: 22321215. DOI: 10.1016/j.bmcl.2012.01.053
- Choong CJ, Sasaki T, Hayakawa H, et al. A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson's disease. Neurobiol Aging. 2016; 37: 103-116. PMID: 26545632. DOI: 10.1016/j.neurobiolaging.2015.10.001
- Cacan E, Ali MW, Boyd NH, et al. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLOS ONE. 2014; 9(1): 1-11. PMID: 24475290. DOI: 10.1371/journal.pone.0087455
- Sarkar S, Abujamra AL, Loew JE, et al. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011; 31(9): 2723-2732. PMID: 21868513
- Frew AJ, Lindemann RK, Martin BP, et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci USA. 2008; 105(32): 11317-11322. PMID: 18685088. DOI: 10.1073/pnas.0801868105
- Sarkar S, Faller DV. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleotides. 2011; 21(1): 47-53. PMID: 21281128. DOI: 10.1089/oli.2010.0259
- Mataga MA, Rosenthal S, Heerboth S, et al. Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res. 2012; 32(7): 2523-2529. PMID: 22753709
- Lapinska K, Housman G, Byler S, et al. The Effects of Histone Deacetylase Inhibitor and Calpain Inhibitor Combination Therapies on Ovarian Cancer Cells. Anticancer Res. 2016; 36(11): 5731-5742. PMID: 27793894. DOI: 10.21873/anticanres.11156
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014; 13(5): 337-356. PMID: 24751816. DOI: 10.1038/nrd4286
- Shimamura T, Chen Z, Soucheray M, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non–small cell lung cancer. Clin. Cancer Res. 2013; 19(22): 6183-6192. PMID: 24045185. DOI: 10.1158/1078-0432.CCR-12-3904
- Baratta MG, Schinzel AC, Zwang Y, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci USA. 2015; 112(1): 232-237. PMID: 25535366. DOI: 10.1073/pnas.1422165112
- Kurimchak AM, Shelton C, Duncan KE, et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 2016; 16(5): 1273-1286. PMID: 27452461. DOI: 10.1016/j.celrep.2016.06.091
- Zhang Z, Ma P, Jing Y, et al. BET bromodomain inhibition as a therapeutic strategy in ovarian cancer by downregulating FoxM1. Theranostics. 2016; 6(2): 219-230. PMID: 26877780. DOI: 10.7150/thno.13178
- Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016; 529(7586): 413-417. PMID: 26735014. DOI: 10.1038/nature16508
- Mack GS. Epigenetic cancer therapy makes headway. J Natl Cancer Inst Monogr. 2006; 98(20): 1443-1444. PMID: 17047192. DOI: 10.1093/jnci/djj447
- Arce C, Segura-Pacheco B, Perez-Cardenas E, et al. Hydralazine target: From blood vessels to the epigenome. J Transl Med. 2006; 4(10): 1-16. PMID: 16507100. DOI: 10.1186/1479-5876-4-10
- Candelaria M, Gallardo-Rincón D, Arce C, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol. 2007; 18(9): 1529-1538. PMID: 17761710. DOI: 10.1093/annonc/mdm204
- Phiel CJ, Zhang F, Huang EY, et al.: Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001; 276(39): 36734-36741. PMID: 11473107. DOI: 10.1074/jbc.M101287200
- Priya R, Ilavenil S, Kaleeswaran B, et al. Effect of Lawsonia inermis on tumor expression induced by Dalton’s lymphoma ascites in Swiss albino mice. SAUDI J BIOL SCI. 2011; 18(4): 353-359. PMID: 23961147. DOI: 10.1016/j.sjbs.2011.04.001
- Singh DK, Luqman S. Lawsonia inermis (L.): A perspective on anticancer potential of Mehndi/Henna. Biomedical Research and Therapy. 2014; 1(4): 112-120.
- Sharma RD, Raghuram TC, Rao NS. Effect of fenugreek seeds on blood glucose and serum lipids in type I diabetes. Eur J Clin Nutr. 1990; 44(4): 301-306. PMID: 2194788
- Khoja KK, Shaf G, Hasan TN, et al. Fenugreek, a naturally occurring edible spice, kills MCF-7 human breast cancer cells via an apoptotic pathway. Asian Pac J Cancer Prev. 2011; 12(12): 3299-3304. PMID: 22471470
- Losso JN, Holliday DL, Finley JW, et al. Fenugreek bread: a treatment for diabetes mellitus. J Med Food. 2009; 12(5): 1046-1049. PMID: 19857068. DOI: 10.1089/jmf.2008.0199
- Dirar AI, Mohamed MA, Osman WJ, et al. A Phytopharmacological review on four antitumor medicinal plants grown in sudan. Am J PharmTech Res. 2014; 4(5): 28-41.
- Liu CM, Sun YZ, Sun JM, et al. Protective role of quercetin against lead-induced inflammatory response in rat kidney through the ROS-mediated MAPKs and NF-κB pathway. Biochim Biophys Acta Gen Subj. 2012; 1820(10): 1693-1703. PMID: 22728154. DOI: 10.1016/j.bbagen.2012.06.011
- Vijayababu MR, Arunkumar A, Kanagaraj P, et al. Quercetin downregulates matrix metalloproteinases 2 and 9 proteins expression in prostate cancer cells (PC-3). Mol Cell Biochem. 2006; 287(1-2): 109-116. PMID: 16645725. DOI: 10.1007/s11010-005-9085-3
- Yamazaki S, Miyoshi N, Kawabata K, et al. Quercetin-3-O-glucuronide inhibits noradrenaline-promoted invasion of MDA-MB-231 human breast cancer cells by blocking β2-adrenergic signaling. Arch Biochem Biophys. 2014; 557(2014): 18-27. PMID: 24929186. DOI: 10.1016/j.abb.2014.05.030
- Toyota M, Issa JP. Epigenetic changes in solid and hematopoietic tumors. Semin Oncol. 2005; 32(5): 521-530. PMID: 16210093. DOI: 10.1053/j.seminoncol.2005.07.003
- Issa JP. DNA methylation as a therapeutic target in cancer. Clin Cancer Res. 2007; 13(6): 1634-1637. PMID: 17363514. DOI: 10.1158/1078-0432.CCR-06-2076
- Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene. 2002; 21(2002): 5483–5495. PMID: 12154409. DOI: 10.1038/sj.onc.1205699
- Gaudet F, Hodgson JG, Eden A, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003; 300(5618): 489-492. PMID: 12702876. DOI: 10.1126/science.1083558
- Brueckner B, Boy RG, Siedlecki P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005; 65(14): 6305-6311. PMID: 16024632. DOI: 10.1158/0008-5472.CAN-04-2957
- Messina M, Nagata C, Wu AH. Estimated Asian adult soy protein and isoflavone intakes. Nutr Cancer. 2006; 55(1): 1-2. PMID: 16965235. DOI: 10.1207/s15327914nc5501_1
- Ahn MY, Kang DO, Na YJ, et al. Histone deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell migration via class II histone deacetylase 4 silencing. Cancer lett. 2012; 201325(2): 189-199. PMID: 22781396. DOI: 10.1016/j.canlet.2012.06.017
- Fang F, Munck J, Tang J, et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin Cancer Res. 2014; 20(24): 6504-6516. PMID: 25316809. DOI: 10.1158/1078-0432.CCR-14-1553
- Manetta A, Blessing JA, Look KY. A phase II study of fazarabine in patients with advanced ovarian cancer. A Gynecologic Oncology Group study. Am J Clin Oncol. 1995; 18(2): 156-157. PMID: 7534978
- Ravindran J, Prasad S, Aggarwal BB. Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J. 2009; 11(3): 495-510. PMID: 19590964. DOI: 10.1208/s12248-009-9128-x
- Afroze SH, Peddaboina C, McDowell AB, et al. Differential effects of in vitro treatment with cinobufotalin on three types of ovarian cancer cells. Anticancer Res. 2018; 38(10): 5717-5724. PMID: 30275192. DOI: 10.21873/anticanres.12909
- Yamauchi K, Afroze SH, Mitsunaga T, et al. 3, 4’, 7-O-Trimethylquercetin inhibits invasion and migration of ovarian cancer cells. Anticancer Res. 2017; 37(6): 2823-2829. PMID: 28551617. DOI: 10.21873/anticanres.11633
- Ashraf AZ, Afroze SH, Yamauchi K, et al. Differential mechanism of action of 3, 4’, 7-O-trimethylquercetin in three types of ovarian cancer cells. Anticancer Res. 2018; 38(9): 5131-5137. PMID: 30194160. DOI: 10.21873/anticanres.12835
- Gozum S, Tezel A, Koc M. Complementary alternative treatments used by patients with cancer in eastern Turkey. Cancer Nurs. 2003; 26(3): 230-236. PMID: 12832956
- Coverdale JP, Romero-Canelón I, Sanchez-Cano C, et al. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat Chem. 2018; 10(3): 347-354. PMID: 29461524. DOI: 10.1038/nchem.2918
- Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016; 41(3): 211-218. PMID: 26778478. DOI: 10.1016/j.tibs.2015.12.001
- Liu Z, Habtemariam A, Pizarro AM, et al. Organometallic half-sandwich iridium anticancer complexes. Open J Med Chem. 2011; 54(8): 3011-3026. PMID: 21443199 DOI: 10.1021/jm2000932
- Wargovich MJ, Woods C, Hollis DM, et al. Herbals, cancer prevention and health. J Nutr. 2001; 131(11): 3034-3036. PMID: 11694643. DOI: 10.1093/jn/131.11.3034S
- Florea AM, Büsselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers. 2011; 3(1): 1351-1371. PMID: 24212665. DOI: 10.3390/cancers3011351