
Hypoxia-Induced Autophagy Degrades Stromal Lumican into Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma: A Mini-Review
Bhaswati Sarcar1, Xinqun Li2, Jason B. Fleming1*
1Department of Gastrointestinal Oncology, H. Lee Moffitt Cancer Center and Research Institute, FL, USA
2Department of Surgical Oncology, The University of Texas MD Anderson Cancer Center, TX, USA
Abstract
The extracellular matrix (ECM) in the tumor microenvironment (TME) has gained considerable interest in recent years as a crucial component in fundamental cellular processes and provides novel therapeutic targets. Lumican is a class II small leucine-rich proteoglycan with a key role in ECM organization and modulation of biological functions dependent on tumor type, abundance, and stage of disease. The presence of stromal lumican in the ECM surrounding pancreatic ductal adenocarcinoma (PDAC) inhibits cancer cell replication and is associated with improved patient outcomes after multimodal therapies. In this mini-review, we re-present our novel findings describing how hypoxia (1% O2) within the TME influences stromal lumican expression and secretion. We observed that hypoxia specifically inhibited lumican expression and secretion post-transcriptionally only from pancreatic stellate cells. Hypoxia-induced increased lactate production did not influence lumican expression. Notably, autophagy was induced by hypoxia in ex vivo cultures of patient-derived primary PDAC xenograft and pancreatic stellate cells; however, the cancer cells remain unaffected. Moreover, hypoxia-inducible factor (HIF)-1α expression or inhibition of AMP-regulated protein kinase (AMPK) activation within hypoxic stellate cells restored lumican expression levels. Interestingly, AMPK inhibition attenuated hypoxia-reduced phosphorylation of the mTOR/p70S6K/4EBP signaling pathway. The aim of this mini-review is to summarize our recent publication that hypoxia reduces stromal lumican in PDAC through autophagy-mediated degradation and reduction in protein synthesis within pancreatic cancer stellate cells. This may provide another plausible mechanism by which hypoxia-induced stromal autophagy leads to cancer growth.
Pancreatic ductal adenocarcinoma and Lumican
Pancreatic ductal adenocarcinoma (PDAC) is a very aggressive malignancy with a high incidence of distant metastasis1 and is projected to be the second leading cause of cancer death by 20302. The pathways that promote invasion and metastasis of PDAC are elusive. However, elucidating the mechanisms behind the metastatic efficiency of this deadly disease is important to develop a novel therapeutic approach. PDAC exhibits extensive desmoplastic stroma, which derives from activated pancreatic stellate cells. This activation results in proliferation, leading to the production of collagen, laminin, fibronectin3, and lumican4 within the extracellular matrix (ECM). Lumican, a member of a class II family of small leucine-rich proteoglycans, is highly upregulated in different cancer types, especially pancreas, breast, and cervical cancer5-8, and affects the proliferation, migration, and adhesion of cancer cells through a variety of mechanisms4,9-13. Studies have demonstrated differential distribution of lumican between cancer cells and the reactive tumor stroma4,13-16. In pancreatic cancer, lumican is specifically localized in alpha cells of islets, acinar cells, collagen fibrils, fibroblasts close to pancreatic cancer cells, and cancer cells5. Our previous report and others’ confirm that pancreatic stellate cells are the major source of lumican production and secretion4,6. Recently, it has been shown that heat shock protein 47 (HSP47) interacts with and promotes the secretion of lumican in the ECM17. Lumican was shown to influence cell function through various mechanisms in a tissue-specific manner in different cancers. In the tumor microenvironment (TME) of primary PDAC, the presence of lumican within the ECM has been positively and negatively correlated with tumor progression. It has previously been reported that the localization of lumican in the stromal tissue correlates with advanced-stage pancreatic cancer, retroperitoneal and duodenal invasion, and tends to correlate with shorter survival18. Degradation of lumican by matrix metalloproteinases MTI-MMP resulted in the abrogation of p21-mediated suppression of tumorigenicity by lumican19. On the other hand, a recent study has demonstrated that glycosylated lumican possesses anti-tumor activity by directly interacting with the catalytic domain of MMP-14 and inhibiting its activity20. It was reported that lumican manifested anticancer activity in invasive breast cancer cells by modifying cell morphology, evoking EMT/MET reprogramming, and suppressing matrix metalloproteinases and epidermal growth factor receptors (EGFRs)21. Conversely, in the gastric cancer–associated fibroblasts, lumican activated the β1 integrin–mediated FAK signaling, thereby enhancing tumor growth22. Moreover, secreted 70-kDa lumican by PDAC cells stimulated growth and inhibited invasion of human pancreatic cancer. It also activated ERK signaling, induced α3 integrin expression, and decreased active matrix metalloproteinase-923. On the contrary, lumican overexpression within the pancreatic cancer TME in vivo produced uniformly smaller tumors, correlated with reduced vascular density, enhanced Fas- mediated endothelial apoptosis, and reduced angiogenesis in TME24,25. A key finding in our previous study was that the activated pancreatic stellate cells within the TME are the principle source of stromal lumican, and the presence of lumican within the stroma of a primary PDAC tumor is associated with decreased metastasis and prolonged survival in early-stage pancreatic cancer12,13. Mechanistically, we demonstrated that the activated pancreatic stellate cells within PDAC secrete lumican under the negative control of TGF-β and enhance stellate cell adhesion and mobility in a collagen-rich environment4. We also demonstrated that extracellular lumican physically binds with EGFRs to trigger EGFR internalization, downregulation of EGFR, and its downstream signal. This resulted in apoptosis, glycolytic metabolism inhibition, and entry into a quiescent state12,26. Mounting evidence reported in multiple studies on PDAC by our group4,12,13,26 and several others using different cancer models15,27-30 suggests that lumican is a key regulator of ECM and ECM-cell interactions within the TME. Overall, lumican is shown to have an anti-tumor role in a context-dependent manner, and the conflicting results regarding the precise role of lumican in tumor progression and regression depend on the specific correlation between lumican tissue-specific abundance, distribution, and stage of disease in PDAC.
Hypoxia Attenuates the Secretion of Lumican by Pancreatic Stellate Cells
The PDAC TME is characterized by dense desmoplastic stroma and resultant hypoxia, which also drives angiogenesis, immune suppression, and numerous signaling events that promote cancer progression, metastasis, and poor patient survival31-34. The influence of hypoxia on stromal composition and secreted extracellular protein such as lumican is not clearly understood. Therefore, in our recent publication35, our major focus was to investigate the extent to which hypoxia influences stromal expression and secretion of lumican in PDAC. We observed that hypoxia significantly reduced lumican expression and secretion from pancreatic stellate cells but not cancer cells. Our results revealed that hypoxia stimulates HIF-1α and AMP-regulated protein kinase (AMPK) activation, which leads to autophagy and subsequent reduction in cellular and secreted lumican (Figure 1). We found that AMPK activation blocked the hypoxia-activated mTOR signaling pathway, which in turn inhibited lumican protein synthesis (Figure 1). Therefore, the hypoxia-activated cell signaling event reduces the level of lumican production by stellate cells in the ECM of PDAC.
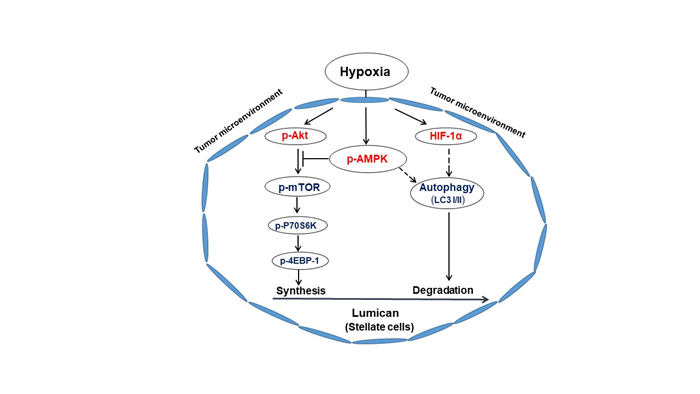
Desmoplastic fibrosis in PDAC tumors inhibits the delivery of nutrients and oxygen, which produces a TME that is characterized by hypoxia and metabolic stress. We expanded our study35 by applying a robust ex vivo organotypic live-tissue culture system to mimic tumor-stroma coevolution and provide a hypoxic environment that mimics the TME. The culture system consisted of precision-cut and uniform small tissue slices (200µm) derived from patient-derived primary PDAC xenograft (PDX) tumors. Importantly, the tissue slices remained viable for at least 5 days. In our previous study, we have shown that microenvironment components, tumor architecture, and cell signaling pathways36 are well preserved in those tumor tissue slices. The slices were cultured in hypoxia (1% O2) for up to 48 hrs. and this unique ex vivo system allowed for the measurement of autophagic changes within the stroma and the reduction of stromal lumican that is exposed to hypoxic conditions. We went on to confirm the finding that hypoxia induces autophagy and attenuates lumican secretion by conducting in vitro studies using activated human stellate cells (HPSCs and HPaSTeC) cultured from human PDAC tumors.
Hypoxia-Induced Autophagy-Mediated Degradation of Lumican in Tumor Stroma
Cellular protein degradations are governed by 2 major pathways in the eukaryotic system: the ubiquitin-proteosome system (UPS) and autophagy. The latter is primarily responsible for the degradation of aggregated proteins and cellular organelles37. The hypoxia and metabolic stress within the TME aggravated cellular autophagy. Previous autophagy-related studies focused only on cancer cells38,39 but recent advancements in research on autophagy have led to a focus on the tumor stroma, preferably referred to as the “autophagic tumor stroma,” which is an adaptation process where the autophagy of stromal cells provides fuel for cancer cells within the TME39-41. Next, to determine which pathway is involved in the lumican degradation process, stellate cells were treated with a UPS inhibitor and autophagy-lysosomal inhibitors in the normoxic and hypoxic environments. Our data indicated that hypoxiareduced stromal lumican via autophagy, not the UPS pathway. We went on to confirm this finding by knocking down proautophagic signaling gene Beclin 1 (Atg6)42 by using RNA interference. Silencing of Beclin 1 set back the hypoxia-induced decrease in lumican that is noticed in stellate cells. We also confirmed the cellular colocalization of the lumican and lysosomal marker LAMP in hypoxic stellate cells. This indicated that lumican is predominantly transported to the lysosome when stellate cells are hypoxic. It is well accepted that hypoxia initiates tumor cell autophagy in different cancers43-45. In our recent study35, we sought to determine whether hypoxia promotes autophagy within pancreatic stellate cells, and our investigation resulted in increasing expressions of the canonical autophagic marker LC3 in both stellate cells and PDX slices that were cultured selectively in hypoxic conditions. Our observations suggested that hypoxia induces autophagy, which results in the degradation of lumican within PDAC stellate cells.
Activated AMPK Negatively Regulates Hypoxia-Induced Stromal Lumican Synthesis
HIF-1α and the proautophagic module AMPK have been known to influence hypoxia-induced autophagy46-48. Inquisitively, we have investigated their role in mediating hypoxia-induced autophagy in pancreatic stromal cells. We evaluated the colocalizations of lumican with HIF-1α and phosphorylated AMPK at the interface between cancer cells and stromal cells in patient tumor tissues. Hypoxia stimulated AMPK activation and the increased expressions of HIF-1α and LC3 in vitro and ex vivo. Treatments consisting of pharmacological AMPK inhibitors and siRNA knockdown of HIF-1α both inhibited hypoxia-induced reduction of lumican expressions. Intriguingly, to define the role of HIF-1α, we transfected the stellate cells with wild type and mutant HIF-1α construct. We noticed that the native HIF-1α, not the mutant, is required for lumican degradation in stromal cells. Therefore, hypoxia-related HIF-1α and AMPK activation mediated the induction of autophagy in the stroma, resulting in lumican degradation. Mechanistically, we found that hypoxia does not influence lumican protein synthesis transcriptionally. We next targeted PI3K/Akt/mTOR pathways and their downstream signaling in stromal cells and in PDX slices in hypoxia to examine the involvement of translational machinery related to lumican protein synthesis. Interestingly, Akt phosphorylation was significantly increased, whereas its remaining antiautophagic node signals, mTOR, P70S6K, and 4-EBP-1 (Figure 1), were decreased during exposure to hypoxia. This finding is concomitant with the hypoxia-enhanced AMPK activation. Previous literature has suggested that Akt-mediated mTOR signaling can be blocked by activated pro-autophagic signaling molecule AMPK, which phosphorylates TSC249,50. Interestingly, treatment of stellate cells with the AMPK inhibitor reversed the hypoxia-related reduction in the mTOR pathway and its downstream signaling (Figure 1). Moreover, it also enhanced the lumican expression transcriptionally, which indicated that hypoxia-induced mTOR activation was blocked by activated AMPK. Therefore, molecular mechanistic studies by our group demonstrated that activated AMPK in hypoxic stellate cells negatively impacts lumican protein synthesis.
Downregulation of Stromal Lumican is not Influenced by Increased Lactate Production
Nearly a century ago, Warburg and his colleague made the seminal observation that the common feature of cancer cell metabolism is increased glucose uptake and the fermentation of glucose into lactate to promote growth, survival, proliferation, and long-term maintenance51. The lactate can be transferred to other cells through specialized transporters, which provides another fuel source for oxidative mitochondrial metabolism52,53. Recently, stromal cells have been recognized as a lactate source for use by cancer cells54. Therefore, in this context, we examined whether stromal lactate, triggered by hypoxia, influenced the observed reduction of lumican in TME. We found that the presence or absence of lactate did not influence lumican expression, although hypoxia dramatically enhanced the lactate dehydrogenase A (LDHA) expression and secretion of lactate in stromal cells. Our future study will assess the expression of a lactate transporter, monocarboxylate transporter 1 (MCT1), and the activation of AMPK and HIF-1α in PDAC stromal cells to gain a better insight into lactate shuttling from PDAC to its stromal cells.
Conclusions
This review summarizes our recent publication35 addressing the factors within the TME that influence lumican production in stromal cells. We consistently found that 1) hypoxia-induced autophagy in PDAC stellate cells; 2) subsequent lumican degradation occurred exclusively in stromal cells, leaving the cancer cells, indicating that stellate cells are more sensitive to hypoxia with respect to lumican levels; and 3) stromal autophagy decreased stromal lumican secretion, which was linked to tumor growth12,13. In conclusion, these recent findings by our group provide important new insights to build and deepen our understanding of the molecular interactions between stromal lumican and the TME and provide a rationale to design credible antitumor approaches against pancreatic cancer.
Acknowledgments
We thank Paul Fletcher (Moffitt Cancer Center) for editorial assistance. The study was supported by grants from the Skip Viragh Family and W. Smith Foundations, National Institute of Health grant T32CA009599, an in-kind grant from the Center for Advanced Biomedical Imaging and GE Healthcare, and U54 CA210181-01 “Center for Immunotherapeutic Transport Oncophysics (CITO)” grant.
References
- Iozzo RV. The family of the small leucine-rich proteoglycans: key regulators of matrix assembly and cellular growth. Crit Rev Biochem Mol Biol. 1997; 32(2): 141-174.
- Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74(11): 2913-2921.
- Apte MV, Wilson JS, Lugea A, et al. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013; 144(6): 1210-1219.
- Kang Y, Roife D, Lee Y, et al. Transforming Growth Factor-beta Limits Secretion of Lumican by Activated Stellate Cells within Primary Pancreatic Adenocarcinoma Tumors. Clin Cancer Res. 2016; 22(19): 4934-4946.
- Ping Lu Y, Ishiwata T, Asano G. Lumican expression in alpha cells of islets in pancreas and pancreatic cancer cells. J Pathol. 2002; 196(3): 324-330.
- Koninger J, Giese T, di Mola FF, et al. Pancreatic tumor cells influence the composition of the extracellular matrix. Biochem Biophys Res Commun. 2004; 322(3): 943-949.
- Leygue E, Snell L, Dotzlaw H, et al. Expression of lumican in human breast carcinoma. Cancer Res. 1998; 58(7): 1348-1352.
- Naito Z, Ishiwata T, Kurban G, et al. Expression and accumulation of lumican protein in uterine cervical cancer cells at the periphery of cancer nests. Int J Oncol. 2002; 20(5): 943-948.
- D'Onofrio MF, Brezillon S, Baranek T, et al. Identification of beta1 integrin as mediator of melanoma cell adhesion to lumican. Biochem Biophys Res Commun. 2008; 365(2): 266-272.
- Seomun Y, Joo CK. Lumican induces human corneal epithelial cell migration and integrin expression via ERK 1/2 signaling. Biochem Biophys Res Commun. 2008; 372(1): 221-225.
- Kang Y, Zhang R, Suzuki R, et al. Two-dimensional culture of human pancreatic adenocarcinoma cells results in an irreversible transition from epithelial to mesenchymal phenotype. Lab Invest. 2015; 95(2): 207-222.
- Li X, Kang Y, Roife D, et al. Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene. 2017; 36(38): 5432-5438.
- Li X, Truty MA, Kang Y, et al. Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin Cancer Res. 2014; 20(24): 6529-6540.
- Nikitovic D, Katonis P, Tsatsakis A, et al. Lumican, a small leucine-rich proteoglycan. IUBMB Life. 2008; 60(12): 818-823.
- Brezillon S, Pietraszek K, Maquart FX, et al. Lumican effects in the control of tumour progression and their links with metalloproteinases and integrins. FEBS J. 2013; 280(10): 2369-2381.
- Wegrowski Y, Maquart FX. Involvement of stromal proteoglycans in tumour progression. Crit Rev Oncol Hematol. 2004; 49(3): 259-268.
- Ishikawa Y, Rubin K, Bachinger HP, et al. The endoplasmic reticulum-resident collagen chaperone Hsp47 interacts with and promotes the secretion of decorin, fibromodulin and lumican. J Biol Chem. 2018.
- Ishiwata T, Cho K, Kawahara K, et al. Role of lumican in cancer cells and adjacent stromal tissues in human pancreatic cancer. Oncol Rep. 2007; 18(3): 537-543.
- Li Y, Aoki T, Mori Y, et al. Cleavage of lumican by membrane-type matrix metalloproteinase-1 abrogates this proteoglycan-mediated suppression of tumor cell colony formation in soft agar. Cancer Res. 2004; 64(19): 7058-7064.
- Pietraszek-Gremplewicz K, Karamanou K, Niang A, et al. Small leucine-rich proteoglycans and matrix metalloproteinase-14: Key partners? Matrix Biol. 2019; 75-76: 271-285.
- Karamanou K, Franchi M, Piperigkou Z, et al. Lumican effectively regulates the estrogen receptors-associated functional properties of breast cancer cells, expression of matrix effectors and epithelial-to-mesenchymal transition. Sci Rep. 2017; 7: 45138.
- Wang X, Zhou Q, Yu Z, et al. Cancer-associated fibroblast-derived Lumican promotes gastric cancer progression via the integrin beta1-FAK signaling pathway. Int J Cancer. 2017; 141(5): 998-1010.
- Yamamoto T, Matsuda Y, Kawahara K, et al. Secreted 70kDa lumican stimulates growth and inhibits invasion of human pancreatic cancer. Cancer Lett. 2012; 320(1): 31-39.
- Williams KE, Fulford LA, Albig AR. Lumican reduces tumor growth via induction of fas-mediated endothelial cell apoptosis. Cancer Microenviron. 2010; 4(1): 115-126.
- Sharma B, Ramus MD, Kirkwood CT, et al. Lumican exhibits anti-angiogenic activity in a context specific manner. Cancer Microenviron. 2013; 6(3): 263-271.
- Li X, Roife D, Kang Y, et al. Extracellular lumican augments cytotoxicity of chemotherapy in pancreatic ductal adenocarcinoma cells via autophagy inhibition. Oncogene. 2016; 35(37): 4881-4890.
- Brezillon S, Radwanska A, Zeltz C, et al. Lumican core protein inhibits melanoma cell migration via alterations of focal adhesion complexes. Cancer Lett. 2009; 283(1): 92-100.
- Brezillon S, Venteo L, Ramont L, et al. Expression of lumican, a small leucine-rich proteoglycan with antitumour activity, in human malignant melanoma. Clin Exp Dermatol. 2007; 32(4): 405-416.
- Brezillon S, Zeltz C, Schneider L, et al. Lumican inhibits B16F1 melanoma cell lung metastasis. J Physiol Pharmacol. 2009; 60 Suppl 4: 15-22.
- Gore J, Korc M. Pancreatic cancer stroma: friend or foe? Cancer Cell. 2014; 25(6): 711-712.
- Buchler P, Reber HA, Lavey RS, et al. Tumor hypoxia correlates with metastatic tumor growth of pancreatic cancer in an orthotopic murine model. J Surg Res. 2004; 120(2): 295-303.
- Bao B, Ali S, Ahmad A, et al. Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and miR-21, which can be attenuated by CDF treatment. PLoS One. 2012; 7(12): e50165.
- Farazi TA, Spitzer JI, Morozov P, et al. miRNAs in human cancer. J Pathol. 2011; 223(2): 102-115.
- Greither T, Grochola LF, Udelnow A, et al. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer. 2010; 126(1): 73-80.
- Li X, Lee Y, Kang Ya, et al. Hypoxia-induced autophagy of stellate cells inhibits expression and secretion of lumican into microenvironment of pancreatic ductal adenocarcinoma. Cell Death & Differentiation. 2018.
- Roife D, Dai B, Kang Y, et al. Ex Vivo Testing of Patient-Derived Xenografts Mirrors the Clinical Outcome of Patients with Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2016; 22(24): 6021-6030.
- Lilienbaum A. Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol. 2013; 4(1): 1-26.
- Apel A, Zentgraf H, Buchler MW, et al. Autophagy-A double-edged sword in oncology. Int J Cancer. 2009; 125(5): 991-995.
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010; 12(9): 814-822.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132(1): 27-42.
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004; 306(5698): 990-995.
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010; 12(9): 823-830.
- Cao Y, Luo Y, Zou J, et al. Autophagy and its role in gastric cancer. Clin Chim Acta. 2018; 489: 10-20.
- Zhang J, Chu D, Kawamura T, et al. GRIM-19 repressed hypoxia-induced invasion and EMT of colorectal cancer by repressing autophagy through inactivation of STAT3/HIF-1alpha signaling axis. J Cell Physiol. 2018.
- Huang S, Qi P, Zhang T, et al. The HIF1alpha/miR2243p/ATG5 axis affects cell mobility and chemosensitivity by regulating hypoxiainduced protective autophagy in glioblastoma and astrocytoma. Oncol Rep. 2018.
- Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008; 283(16): 10892-10903.
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004; 23(16): 2891-2906.
- Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004; 23(16): 2746-2756.
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009; 37(Pt 1): 217-222.
- Chen W, Pan Y, Wang S, et al. Cryptotanshinone activates AMPK-TSC2 axis leading to inhibition of mTORC1 signaling in cancer cells. BMC Cancer. 2017; 17(1): 34.
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927; 8(6): 519-530.
- Gladden LB. A lactatic perspective on metabolism. Med Sci Sports Exerc. 2008; 40(3): 477-485.
- Brooks GA. Lactate: link between glycolytic and oxidative metabolism. Sports Med. 2007; 37(4-5): 341-343.
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010; 9(16): 3256-3276.