
Mini-Review: PDPK1 (3-phosphoinositide dependent protein kinase-1), An Emerging Cancer Stem Cell Target
Bogdan Domrachev1, Sitanshu Singh1, Dandan Li2, Udo Rudloff1,2*
1Rare Tumor Initiative, Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
2Thoracic & GI Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
Abstract
Cancer stem cells (CSCs) are subpopulations of tumor cells that possess abilities for self-renewal, differentiation, and tumor initiation. These rare but therapy-recalcitrant cells are assumed to repopulate tumors following administration of systemic chemotherapy driving therapy failure, tumor recurrence, and disease progression. In early clinical trials, anti-CSC therapies have found limited success to-date possibly due to the inherent heterogeneity and plasticity of CSCs and the incomplete characterization of essential CSC targets. Here, we review the role of 3-phosphoinositide dependent protein kinase-1 (PDPK1) as an emerging CSC target. While most previous studies have relied on CSC models which are based on lineage and tissue-specific marker profiles to define the relationships between putative target and CSC traits, this review discusses PDPK1 and its role in CSC biology with an emphasis on CSC systems which are based on proposed function like label-retaining cancer cells (LRCCs).
The cancer stem cell (CSC) hypothesis
The near universal development of resistance to systemic cytotoxic chemotherapy has been a stubbornly incalcitrant problem in oncology for decades. Commonly, after an initial period of tumor regression and disease control patients afflicted by advanced cancers experience disease progression, detoriation of their quality of life, and succumb to their illness1. Thus, the introduction of the CSC hypothesis, which provided a fundamentally novel explanation for the commonly observed treatment failures and raised prospects to overcome therapy resistance via novel therapy approaches, was greeted with great interest2.
Three decades ago, the identification of tumor-initiating cells within hematological malignancies at the Princess Margaret Cancer Centre in Toronto, Canada3,4, and from breast cancer by Clark and colleagues thereafter5, suggested a hierarchical model of carcinogenesis6. Contrary to the clonal evolution model which suggests stochastic cancer cell divisions resulting in biologically equivalent cancer cells in each daughter cell generation, the CSC theory holds that (1) cancer arises from cells with dysregulated self-renewal mechanisms, and (2) cancer is comprised of a heterogeneous mass of cells which include a small fraction of stem-like progenitor cells that drive tumor progression, and that these cells are functionally distinct from the bulk, differentiated cancer cells2,7,8. While the exact origin of the CSC remains debated with experimental research supporting both CSCs arising via malignant transformation from normal stem cells as well as via de-differentiation of cancer cells9,10, the concept of CSCs does explain the inherent heterogeneity of tumors and the commonly observed resistance to chemoradiotherapy2,11 Failure to eradicate these self-renewing, resistant cells leads to repopulation of residual tumors post-chemotherapy treatment with new cancer cells from this pool of cells and is cause for tumor recurrence (Fig. 1A)12. However, accepting an exclusively hierarchical model to capture cancer pathogenesis is at odds with several clinical findings: for most cancers, the concept of effective tumor burden reduction due to the assumed chemosensitivity of the bulk non-CSC tumor cell population is not observed in the clinic as tumor, in particular solid organ malignancies, not uncommonly do not regress at all upon treatment with systemic chemotherapy1. The concept of treatment-escaping CSCs, self-renewal and repopulating proliferating tumor cell progeny does not apply when tumor recurrence occurs very rapidly, or when cancer treatment accelerates tumor growth. The hierarchical model works best in clinical scenarios which initially see a reduction in tumor burden and then, after a latency period, tumors recur.
Mechanisms of cancer stem cell therapy resistance – inherent versus acquired?
A plethora of in vitro, in vivo as well as findings in patients’ tumoral biopsies support that CSCs are inherently, a priori, resistant to chemoradiotherapy8,14. Inherent CSC characteristics mediating resistance to chemoradiotherapy include upregulation of DNA damage repair mechanism on multiple levels including upregulation of G1/M and G2/S checkpoints, efficient scavenging of reactive oxygen species (ROS), increased drug efflux via upregulation of ATP-binding cassettes and other drug transporters, or upregulation of pro-survival, anti-apoptosis regulators like Bcl-2 or Bcl-XL8. Conventional chemotherapies are most effective in rapidly proliferating cells going frequently through S phase. CSCs on the other hand are known to be quiescent cells which have left the cell cycle and thus escape the genotoxic effect of many currently employed chemotherapy agents15,16. Additionally, CSCs can effectively sense chemotherapy agents as xenobiotics and through activation of ALDH1A1 or ALDH3A1 enzymes which effectively can metabolize chemotherapy agents17. However, CSCs are also able to acquire under therapy pressure novel therapy resistance traits18. Following short courses of chemotherapy the fraction of CSCs to non-CSC tumoral bulk cells has been shown to change dramatically14. Comparing CSCs between chemotherapy-naïve versus chemotherapy-resistant conditions, CSCs acquired a more aggressive phenotype manifested by upregulation of the self-renewal factors NANOG, OCT4, and SOX2 as well as of ALDH3A1. Interestingly, effects on these stemness regulators differed between administered chemotherapeutics. The significant plasticity of CSCs adapting their phenotype under chemotherapy pressure is intriguingly also discussed in the origin and initial emerge of CSCs20. Recent findings show that therapeutic pressure can induce in non-CSCs a transient, stem-like state which is drug resistant due to the induction of EMT factors and other CSC traits. These therapy-induced CSC-like cells revert to their original phenotype upon withdrawal of chemotherapy underscoring the dynamic nature of their adaptive mechanisms in response to cancer therapy.
Intratumoral CSC heterogeneity
CSC heterogeneity is, in large, also the result of the complex dynamic interplay and interdependence between CSCs and surrounding stromal milieu forming the CSC niche. One of the best investigated CSC niche–CSC interface are adaptations of CSCs residing in hypoxic areas21. Hypoxic, poorly perfused areas of solid organ cancers are most heavily populated with CSCs. Within these hypoxic niches CSCs create a unique environment by attracting M2-like tumor associated macrophages, suppressing dendritic and cytotoxic T cell function, upregulate inhibitory immune checkpoints or pro-angiogenic signals21,22. CSC residing in peri-vascular regions on the other hand find a nutrient-rich environment, have higher proliferative rates and shape vascularization and ECM formation23. CSCs within the invasive front of tumors have upregulated migratory and invasive capabilities, and the overlap with programs essential for metastasis like e.g. the CXCR4/CXLX12 axis suggests these cells to be causally involved in the metastatic spread of cancers21. Overall, while the ‘niche concept’ is a simplification of the very complex and dynamic interplay of CSCs with nearly all stromal elements ECM components, it provides functional rationale for the phenotypic differences and heterogeneity observed among CSCs. As CSCs need to find their niches, vice versa, they become major educators of their microenvironment. Reciprocally, these educated stromal cells signal back and impact CSC traits like CSC quiescence, self-renewal, or therapy resistance21.
It is also likely that cancer stem cells are coming in and out of the CSC pools increasing heterogeneity and pleiotropism of CSC traits within tumors further9. In this regard, targeting rare, infrequent CSC populations defined by CSC surface markers and an overreliance on such markers to capture CSC populations mediating chemotherapy resistance might have led to the largely disappointing early clinical results of anti-CSC therapy so far7. Thus, there has been a renewed refocus on preclinical CSC models with improved recapitulation of tumoral stem cell biology, like LRCCs, which are less biased by culture conditions, cell lineage markers, or results derived from immune compromised mouse models9.
Here we discuss the role of PDPK1 as an emerging regulator of cancer stemness by reviewing recent findings in functional CSC models like label-retaining cells (LRCCs), studies in autochthonous animal models of cancer, as well as CSC traits supported by PDPK1 (Fig. 1B).
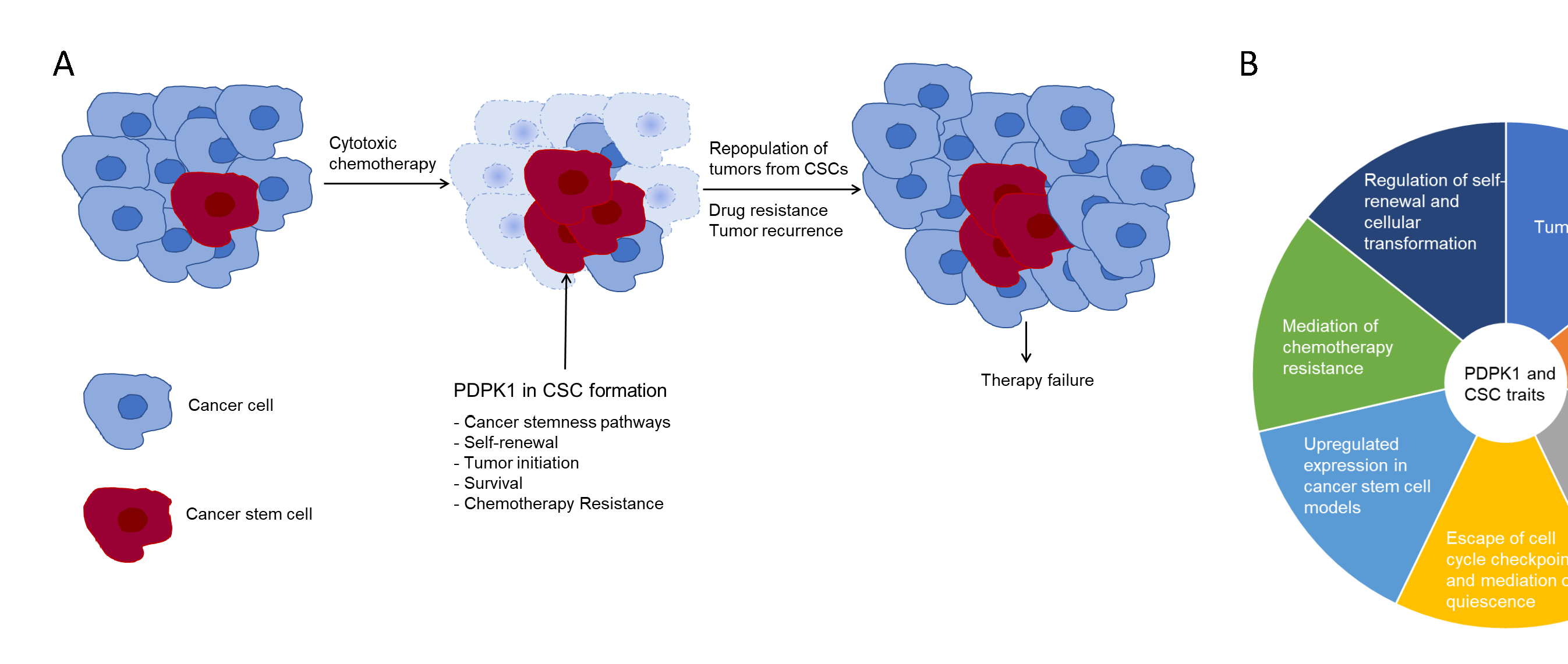
Figure 1: Role of CSCs in mediation of therapy resistance and tumor recurrence. A. Self-renewal of therapy-resistant CSCs repopulate tumors leading to therapy failure including cancer recurrence and disease progression. B. PDPK1 signaling involved in CSC function. CSC traits supported by PDPK1 are listed.
Regulation of cancer stemness pathways by PDPK1
PDPK1 is a phylogenetically conserved member and master regulator of the large AGC kinase family24. PDPK1 possesses a N-terminal kinase and a C-terminal pleckstrin homology (PH) domain which senses PI3K-generated phospho-inositide metabolites, in particular phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3); PIP3), at the inner plasma membrane. The N-terminal kinase domain consists of the activation loop and the PDPK1-interacting fragment (PIF)-pocket which binds to a hydrophobic motif on PDPK1 substrates25. PDPK1 autophosphorylation of the activation loop (at serine 241) activates PDKP126. Signaling output of constitutively activated PDPK1 is primarily determined by the pre-activation state and post-translational modifications of its substates as well as interactions with other protein modulators like the inhibitory 14-3-3 or the tumor suppressor candidate 4 (TUSC4) proteins24,27,28. Whereas AKT activation by PDPK1 is regulated via simultaneous PIP3 binding enhancing proximity and phosphorylation at threonine 308 of the activation loop of AKT, other AGC kinase substrates of PDPK1 like serum and glucocorticoid-regulated kinase 1 (SGK1), p70 ribosomal S6 kinase (p70S6K), protein kinase C (PKC), or p90RSK interact with PDPK1 through their PIF-binding motifs. Post-translational modifications of the PIF domains of these substrate kinases very heavily impact the affinity of PDPK1-kinase interactions and therewith PDPK1 signaling output. Thus the varying pre-activation states of PDPK1 substrate kinases explain the heterogenous and pleiotropic PDPK1 output across different cell lineages and the not uncommonly observed lack of correlation of PDPK1 activity and AKT activation29,30.
Considering the pleiotropism of PDPK1 output, it is not surprising that PDPK1 was found to be an essential regulator of cancer stem cell signal transduction. Best examined is the Hippo signaling pathway31; in its non-activated form, PDPK1 forms in the cytoplasm a complex with Sav1 which allows the Hippo pathway components MST1/2 and LATS to phosphorylate YAP retaining this master regulator in the cytoplasm32,33. Following phosphorylation and membranous recruitment of PDPK1, this complex dissolves, YAP is allowed nuclear entry and activation of the YAP/TAZ stemness program occurs. Constitutively activated PDPK1 has been shown to stimulate β-catenin/Wnt signaling in medulloblastoma where PDPK1 small molecule inhibition increased survival and enhanced the cytotoxic effects of chemotherapeutic drugs34. PDPK1-mediated activation of the AGC kinase S6K1 connects PDPK1 signaling output with the hedgehog pathway in cancer cells35. Recently, PDPK1 has also been shown to function as an upstream regulator of the cancer stemness master regulator Myc36. A summary of PDPK1-mediated regulation of cancer stemness signaling pathways is shown in Fig. 2.
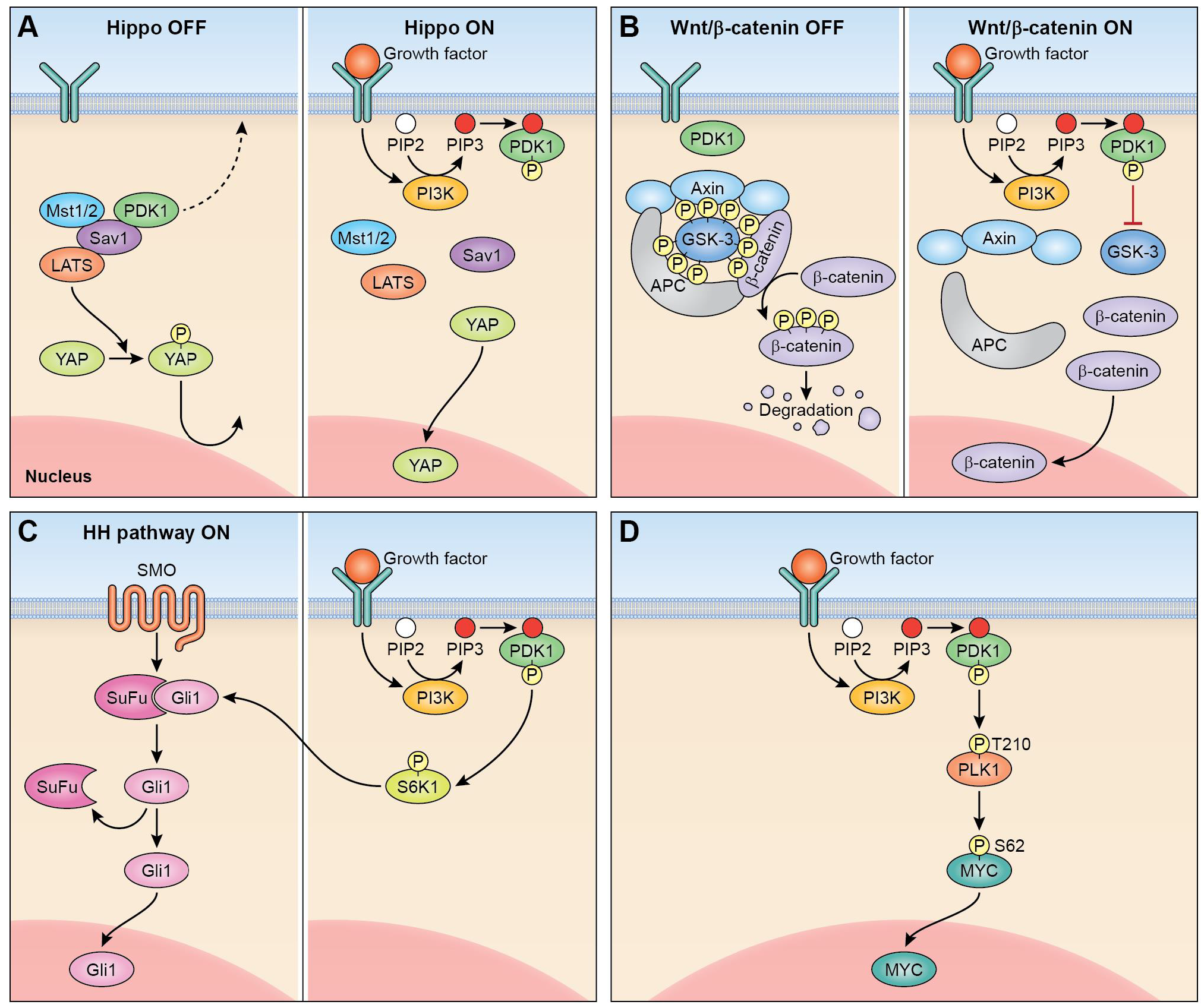
Figure 2: Regulation of cancer stemness pathways by PDPK1. A. Membranous recruitment of PDPK1 (right) activates Hippo pathway. B. Activation of WNT/β-catenin signaling. C. PDPK1 activation of S6 kinase activates hedgehog pathway (HH, hedgehog; SMO, smoothened). D. PDPK1-PLK1-MYC signaling axis in cancer.
Cancer stem cell self-renewal and tumor initiation
PDPK1 has recently been identified as an upstream regulator of the c-Myc oncogene. PDPK1 can phosphorylate PLK1 on threonine 210 which, once phosphorylated, stabilizes Myc from proteasomal degradation via serine 62 phosphorylation36,37. Increased cellular levels of c-Myc promote cancer cell self-renewal, survival, and increased CSC fractions in tumors as loss-of-function experiments resulted in loss of CSCs and CSC functions upon silencing of PDPK136. Of note, PDPK1-governed increased stability of c-Myc was associated with resistance to targeted therapy36,38. That PDPK1-governed CSC function can translate into enhanced tumor initiation in vivo has recently been elegantly shown in autochthonous pancreas cancer models. Eser and colleagues showed that hat PDPK1 is an essential effector of Kras, and that an intact PDPK1/PI3K axis is an essential tumor initiating event in cooperation with KRAS for increased cell plasticity, acinar-to-ductal metaplasia (ADM), and pancreatic ductal adenocarcinoma (PDAC) formation39. Similar findings of PDPK1 promoting tumor initiation in cooperation with Erbb2 or Ras activation have been made in breast cancer cells40,41, or in BRAF V600 mutant melanoma where loss of PDPK1 reduced tumor formation42.
Mediation of resistance to chemotherapy
While there are many studies linking PDPK1 expression level and PDPK1 activation to chemoresistance43, it is not always clear that increased chemoresistance is due to PDPK1-promoted cancer stemness. AKT signaling with its pro-survival, anti-apoptosis functions is, for example, a well-known mediator of chemoresistance in cancer cells independent of tumor cell differentiation state and CSC function. Thus, one of the more compelling examinations of PDPK1-governed cancer stemness mediating resistance to chemotherapy emanates from studies in label-retaining cancer cells (LRCCs). LRCCs are slowly cycling, quiescent cells holding onto intracellular dyes16. Slow cycling LRCCs exhibit cancer stem cell and pluripotency traits and represent a distinct subpopulation of the heterogeneous CSC pool44,45. LRCCs have been shown to be more tumorigenic, mediate therapy resistance, and promote tumor recurrence15,46,47. Treatment with chemotherapy dramatically can increase the fraction of LRCCs48. It is believed that LRCCs undergo asymmetric cell divisions with non-random chromosomal co-segregation45. LRCCs provide a unique opportunity to study cancer stemness within a live, cell-based system that relies on stemness function rather than on marker profiles. Studying label-retaining pancreas cancer cells, Li and coworkers showed that PDPK1 is significantly upregulated in LRCCs compared to the non-LRCC fraction and that PDPK1 regulates survival and response to chemotherapy treatment in LRCCs, a finding also made by another group47,49. Findings that the stemness cell population survives chemotherapy and is the nidus for re-growth and disease relapse have also been made in acute myeloid leukemia (AML) where PDPK1 was found to be a major regulator of leukemia stem cell survival50.
Conclusions and Future Perspectives
PDPK1 is an emerging CSC target in at least some cancer histologies. Therapies aimed at the eradication of CSCs involved in chemoresistance, repopulation of tumors, and cancer recurrence appear to be a logical translation of the cancer stem cell hypothesis into the clinic. While PDPK1 small molecule inhibitors have entered clinical testing it remains to be seen whether the therapeutic window targeting PDPK1 in CSCs versus unwarranted off-target effects on PDPK1 signaling essential in many physiologic processes is large enough for anti-PDPK1 targeted therapy to be safe in patients afflicted by cancer and become clinically feasible.
Disclosures: No conflict of interest.
Financial support:
This project has been funded, in part, with Federal funds from the National Institutes of Health (NIH), and was supported by the Intramural Research Program (IRP) of the NIH, National Cancer Institute, Center for Cancer Research (ZIA BC 011267) as well as donations from ‘Running for Rachel’ and the Pomerenk family via the Rachel Guss and Bob Pomerenk Pancreas Cancer Research Fellowship to NCI.
Disclaimer:
The opinions expressed in this article are the author's own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States Government, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
References
- Blagoev KB, Wilkerson J, Stein WD, et al: Therapies with diverse mechanisms of action kill cells by a similar exponential process in advanced cancers. Cancer Res 74:4653-62, 2014
- Clevers H: The cancer stem cell: premises, promises and challenges. Nat Med 17:313-9, 2011
- Lapidot T, Sirard C, Vormoor J, et al: A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367:645-8, 1994
- Bonnet D, Dick JE: Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3:730-7, 1997
- Al-Hajj M, Wicha MS, Benito-Hernandez A, et al: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983-8, 2003
- Rich JN: Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine (Baltimore) 95:S2-S7, 2016
- Brower V: Cancer Stem Cell Hypothesis Evolves With Emerging Research. J Natl Cancer Inst 108, 2016
- Islam F GV, Lam AK.: Cancer Stem Cells: Role in Tumor Progression and Treatment Resistance. Oncogenomics:77-87, 2019
- Rahman M, Deleyrolle L, Vedam-Mai V, et al: The cancer stem cell hypothesis: failures and pitfalls. Neurosurgery 68:531-45; discussion 545, 2011
- Wang X, Prager BC, Wu Q, et al: Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 22:514-528 e5, 2018
- Saygin C, Matei D, Majeti R, et al: Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 24:25-40, 2019
- Kurtova AV, Xiao J, Mo Q, et al: Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517:209-13, 2015
- Savage P: Chemotherapy curable malignancies and cancer stem cells: a biological review and hypothesis. BMC Cancer 16:906, 2016
- Yu F, Yao H, Zhu P, et al: let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131:1109-23, 2007
- Moore N, Houghton J, Lyle S: Slow-cycling therapy-resistant cancer cells. Stem Cells Dev 21:1822-30, 2012
- Davis JE, Jr., Kirk J, Ji Y, et al: Tumor Dormancy and Slow-Cycling Cancer Cells. Adv Exp Med Biol 1164:199-206, 2019
- Chefetz I, Grimley E, Yang K, et al: A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep 26:3061-3075 e6, 2019
- Das PK, Pillai S, Rakib MA, et al: Plasticity of Cancer Stem Cell: Origin and Role in Disease Progression and Therapy Resistance. Stem Cell Rev Rep 16:397-412, 2020
- Dylla SJ, Beviglia L, Park IK, et al: Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 3:e2428, 2008
- Polyak K, Weinberg RA: Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9:265-73, 2009
- Prager BC, Xie Q, Bao S, et al: Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 24:41-53, 2019
- Zhou W, Ke SQ, Huang Z, et al: Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17:170-82, 2015
- Ghajar CM, Peinado H, Mori H, et al: The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15:807-17, 2013
- Gagliardi PA, Puliafito A, Primo L: PDK1: At the crossroad of cancer signaling pathways. Semin Cancer Biol 48:27-35, 2018
- Biondi RM, Cheung PC, Casamayor A, et al: Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J 19:979-88, 2000
- Casamayor A, Morrice NA, Alessi DR: Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem J 342 ( Pt 2):287-92, 1999
- Sato S, Fujita N, Tsuruo T: Regulation of kinase activity of 3-phosphoinositide-dependent protein kinase-1 by binding to 14-3-3. J Biol Chem 277:39360-7, 2002
- Kurata A, Katayama R, Watanabe T, et al: TUSC4/NPRL2, a novel PDK1-interacting protein, inhibits PDK1 tyrosine phosphorylation and its downstream signaling. Cancer Sci 99:1827-34, 2008
- Vasudevan KM, Barbie DA, Davies MA, et al: AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16:21-32, 2009
- Castel P, Ellis H, Bago R, et al: PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer Cell 30:229-242, 2016
- Pan D: The hippo signaling pathway in development and cancer. Dev Cell 19:491-505, 2010
- Fan R, Kim NG, Gumbiner BM: Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A 110:2569-74, 2013
- Xia H, Dai X, Yu H, et al: EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis 9:269, 2018
- Baryawno N, Sveinbjornsson B, Eksborg S, et al: Small-molecule inhibitors of phosphatidylinositol 3-kinase/Akt signaling inhibit Wnt/beta-catenin pathway cross-talk and suppress medulloblastoma growth. Cancer Res 70:266-76, 2010
- Brechbiel J, Miller-Moslin K, Adjei AA: Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev 40:750-9, 2014
- Tan J, Li Z, Lee PL, et al: PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov 3:1156-71, 2013
- Wang X, Cunningham M, Zhang X, et al: Phosphorylation regulates c-Myc's oncogenic activity in the mammary gland. Cancer Res 71:925-36, 2011
- Cunningham JT, Ruggero D: New connections between old pathways: PDK1 signaling promotes cellular transformation through PLK1-dependent MYC stabilization. Cancer Discov 3:1099-102, 2013
- Eser S, Reiff N, Messer M, et al: Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 23:406-20, 2013
- Maurer M, Su T, Saal LH, et al: 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res 69:6299-306, 2009
- Gagliardi PA, di Blasio L, Orso F, et al: 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia 14:719-31, 2012
- Scortegagna M, Ruller C, Feng Y, et al: Genetic inactivation or pharmacological inhibition of Pdk1 delays development and inhibits metastasis of Braf(V600E)::Pten(-/-) melanoma. Oncogene 33:4330-9, 2014
- Emmanouilidi A, Falasca M: Targeting PDK1 for Chemosensitization of Cancer Cells. Cancers (Basel) 9, 2017
- Hari D, Xin HW, Jaiswal K, et al: Isolation of live label-retaining cells and cells undergoing asymmetric cell division via nonrandom chromosomal cosegregation from human cancers. Stem Cells Dev 20:1649-58, 2011
- Xin HW, Hari DM, Mullinax JE, et al: Tumor-initiating label-retaining cancer cells in human gastrointestinal cancers undergo asymmetric cell division. Stem Cells 30:591-8, 2012
- Xin HW, Ambe CM, Hari DM, et al: Label-retaining liver cancer cells are relatively resistant to sorafenib. Gut 62:1777-86, 2013
- Li D, Mullinax JE, Aiken T, et al: Loss of PDPK1 abrogates resistance to gemcitabine in label-retaining pancreatic cancer cells. BMC Cancer 18:772, 2018
- Schillert A, Trumpp A, Sprick MR: Label retaining cells in cancer--the dormant root of evil? Cancer Lett 341:73-9, 2013
- Dembinski JL, Krauss S: Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis 26:611-23, 2009
- Hu T, Li C, Zhang Y, et al: Phosphoinositide-dependent kinase 1 regulates leukemia stem cell maintenance in MLL-AF9-induced murine acute myeloid leukemia. Biochem Biophys Res Commun 459:692-8, 2015